文獻速遞:頂刊CNS神經(jīng)領域研究新進展11月(下)
小編匯總了2025年11月下旬在Nature、Science、Cell 三大國際頂刊上發(fā)表的神經(jīng)科學領域的研究論文,歡迎閱覽!
想要獲取原文的老師同學們,可以拉到文末,聯(lián)系禮智小客服哦!
- 減輕腦部炎癥與神經(jīng)退行的靶點
- 損傷導致疼痛的產(chǎn)生機制
- 果蠅眼部均勻組織模式形成的機制
- 星形膠質(zhì)細胞如何實現(xiàn)多突觸功能整合?
- Shank3寡聚化調(diào)控突觸后密度凝聚物的材料特性
- 果蠅腿部本體感受的選擇性突觸前抑制
- ABCA7基因變異影響神經(jīng)元磷脂酰膽堿代謝與線粒體功能
- 調(diào)控持續(xù)性疼痛需求狀態(tài)的中樞樞紐是?

2025年11月13日,天津醫(yī)科大學總醫(yī)院/北京天壇醫(yī)院施福東教授課題組在Science期刊上發(fā)表了題名為“Targeting formyl peptide receptor 1 reduces brain inflammation and neurodegeneration”的研究論文,揭示了甲酰肽受體1(FPR1)在推動多發(fā)性硬化(MS)神經(jīng)炎癥與神經(jīng)退行中的核心機制。
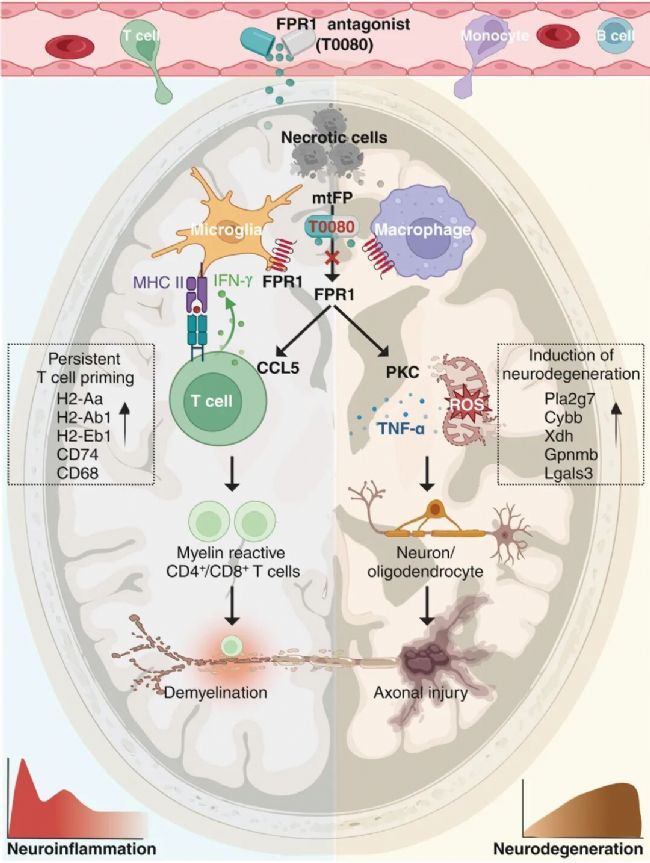
研究發(fā)現(xiàn),MS患者活動性病灶內(nèi)的小膠質(zhì)細胞和巨噬細胞高表達FPR1,且線粒體損傷釋放的內(nèi)源性甲酰肽水平與疾病進展正相關。在實驗模型中,F(xiàn)PR1激活通過蛋白激酶C信號通路觸發(fā)活性氧和腫瘤壞死因子-α的持續(xù)釋放,導致軸突損傷;同時通過分泌CCL5 chemokine促進髓鞘反應性CD4+ T細胞克隆擴增,形成炎癥放大回路。采用中樞神經(jīng)系統(tǒng)滲透性FPR1拮抗劑T0080可有效抑制自身免疫反應并緩解軸突變性。
該研究論證了靶向FPR1信號通路延緩MS進展的治療潛力。
DOI:10.1126/science.adq1177
2、損傷導致疼痛的產(chǎn)生機制

2025年11月20日,美國學者在Science期刊上發(fā)表了題名為“The synaptic ectokinase VLK triggers the EphB2–NMDAR interaction to drive injury-induced pain”的研究論文,研究揭示了細胞外磷酸化在調(diào)控突觸信號傳導與疼痛行為中的新機制。
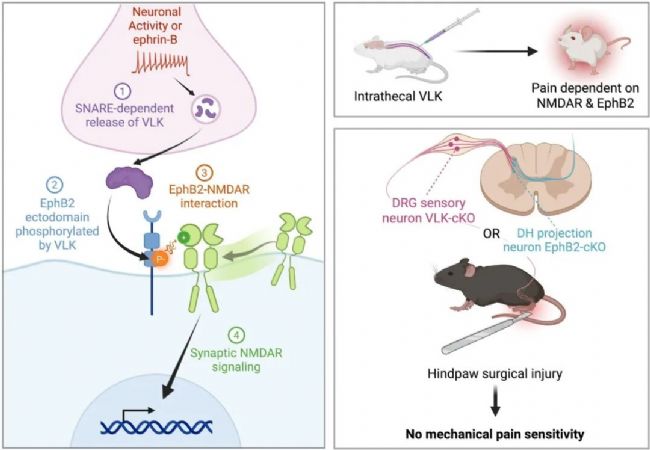
研究發(fā)現(xiàn)分泌型胞外激酶VLK能特異性磷酸化EphB2受體酪氨酸Y504位點,從而誘導EphB2與NMDA受體結合。VLK定位于突觸小泡,在神經(jīng)元活動增強時通過SNARE依賴方式釋放至突觸間隙。在疼痛模型中,感覺神經(jīng)元釋放的VLK通過激活脊髓背角投射神經(jīng)元中的EphB2-NMDAR信號通路,介導機械性痛覺超敏的形成。實驗證實,特異性敲除感覺神經(jīng)元中的VLK或投射神經(jīng)元中的EphB2均能阻斷損傷性疼痛行為,而鞘內(nèi)注射重組VLK則足以誘發(fā)疼痛樣行為。
該研究首次確立了細胞外磷酸化通路在疼痛調(diào)控中的關鍵作用,為靶向突觸外信號治療疼痛提供了新思路。
DOI:10.1126/science.adp1007
3、果蠅眼部均勻組織模式形成的機制

2025年11月20日,美國學者在Science期刊上發(fā)表了題名為“Retinal calcium waves coordinate uniform tissue patterning of the Drosophila eye”的研究論文,研究揭示了果蠅復眼發(fā)育過程中非神經(jīng)元鈣波調(diào)控組織有序化的新機制。
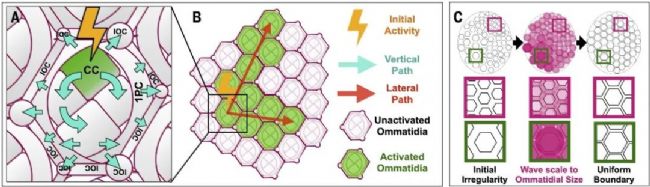
通過新型干透鏡成像技術,研究發(fā)現(xiàn)鈣波由單個小眼內(nèi)錐形細胞通過Cad96Ca受體激活PLC-γ/IP3R信號通路觸發(fā)內(nèi)質(zhì)網(wǎng)鈣釋放。鈣信號通過細胞類型特異性的innexin蛋白間隙連接通道,縱向傳播至小眼內(nèi)所有視網(wǎng)膜細胞(光感受器除外),橫向擴散至相鄰小眼形成跨區(qū)域鈣波。這些鈣波驅(qū)動肌球蛋白II介導的頂端收縮,其信號強度與小眼尺寸成正比,促使前腹側(cè)較大小眼產(chǎn)生更強收縮力,最終消除早期邊界差異,形成均勻有序的視網(wǎng)膜陣列。該機制通過調(diào)整背側(cè)小眼和腹側(cè)小眼的形態(tài)差異,可能滿足其對天空與地面視覺需求的適應性。
研究證實非神經(jīng)元鈣波在感覺組織構建中的關鍵作用,為理解神經(jīng)組織發(fā)育提供新視角。
DOI:10.1126/science.ady5541
4、星形膠質(zhì)細胞如何實現(xiàn)多突觸功能整合?
2025年11月13日,法國、瑞士、意大利、德國、日本以及中國學者聯(lián)合在Cell期刊上發(fā)表了題名為“Astrocytes functionally integrate multiple synapses via specialized leaflet domains”的研究論文,揭示了星形膠質(zhì)細胞葉片狀結構在整合突觸信號中的新機制。
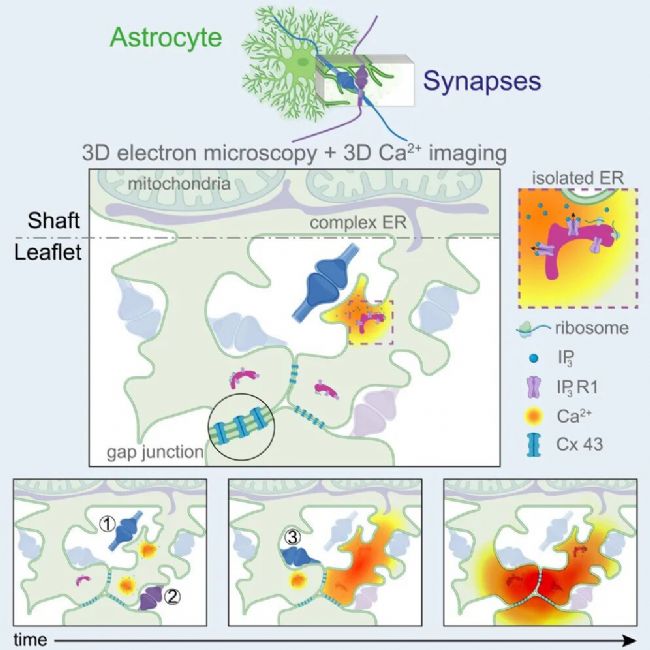
通過高分辨率電鏡與雙光子鈣成像技術,發(fā)現(xiàn)直徑≤250納米的葉片結構包裹著90%的簇狀突觸,其內(nèi)部含有表達IP3R1的微小內(nèi)質(zhì)網(wǎng)囊泡,通過間隙連接形成具有胞質(zhì)連續(xù)性的功能域。研究首次觀測到神經(jīng)元活動可觸發(fā)葉片內(nèi)IP3R1介導的局部鈣信號,這些信號具有獨立起源、融合匯聚的特性,能形成持續(xù)增強的鈣信號平臺。通過同步記錄軸突-葉片鈣活動,證實該復合鈣信號可整合來自不同神經(jīng)元、不同時空尺度的突觸輸入。
研究表明星形膠質(zhì)細胞通過葉片結構的特殊構架與鈣動力學,實現(xiàn)區(qū)別于神經(jīng)元的獨特計算功能,為理解神經(jīng)環(huán)路整合機制提供了新視角。
DOI:10.1016/j.cell.2025.08.036
5、Shank3寡聚化調(diào)控突觸后密度凝聚物的材料特性
2025年11月13日,南方科技大學張明杰院士課題組在Cell期刊上發(fā)表了題名為“Shank3 oligomerization governs material properties of the postsynaptic density condensate and synaptic plasticity”的研究論文,揭示了突觸后密度(PSD)生物凝聚體的材料特性及其在突觸功能中的關鍵作用。
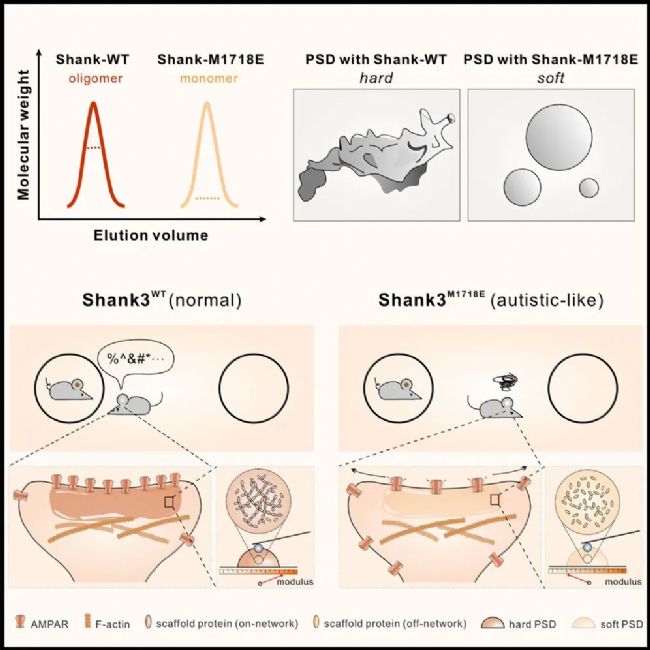
研究發(fā)現(xiàn),通過相分離形成的PSD凝聚體呈現(xiàn)軟玻璃態(tài)材料特性,這種特性源于支架蛋白間特異性多價相互作用形成的網(wǎng)絡滲透。Shank3蛋白的SAM結構域介導的寡聚化對這一網(wǎng)絡構建至關重要,其破壞會導致PSD凝聚體軟化——在Phelan-McDermid綜合征患者中發(fā)現(xiàn)的SHANK3突變即產(chǎn)生此種效應。實驗證實,破壞Shank3寡聚化會削弱PSD網(wǎng)絡滲透,不僅改變凝聚體材料特性,更會損害突觸傳遞與可塑性,引發(fā)小鼠自閉樣行為。
該研究首次確立了生物凝聚體材料特性在學習記憶等神經(jīng)功能中的核心地位,為理解相分離調(diào)控突觸功能的機制提供了新范式。
DOI: 10.1016/j.cell.2025.07.047
6、果蠅腿部本體感受的選擇性突觸前抑制

2025年9月17日,美國學者在Nature期刊上發(fā)表了題名為“Selective presynaptic inhibition of leg proprioception in behaving Drosophila”的研究論文,揭示了果蠅自發(fā)運動過程中特異性抑制本體感覺信號的神經(jīng)環(huán)路機制。
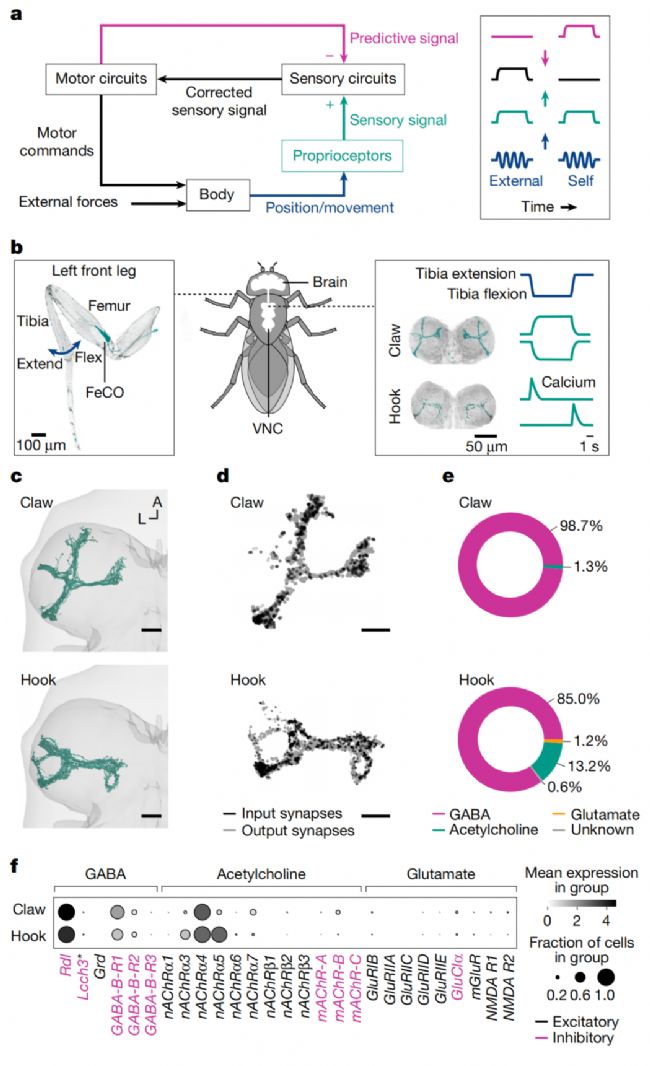
通過行為鈣成像發(fā)現(xiàn),編碼腿部位置的 proprioceptors 在多種行為中持續(xù)活躍,而編碼腿部運動的 proprioceptors 在行走和理毛時被特異性抑制。結合連接組學分析,研究鑒定出一類特定中間神經(jīng)元,通過GABA能突觸前抑制作用于運動編碼型 proprioceptors 的軸突。這些中間神經(jīng)元接收來自并行下行通路的興奮性與抑制性輸入,能以情境特異性和腿部特異性的方式被激活。鈣成像證實該環(huán)路僅在對自主生成的腿部運動(而非被動運動)時激活。
該研究首次揭示了在自發(fā)運動過程中特異性過濾本體感覺信號的神經(jīng)機制,為理解行為依賴的感覺調(diào)控提供了新見解。
DOI:10.1038/s41586-025-09554-2
7、ABCA7基因變異影響神經(jīng)元磷脂酰膽堿代謝與線粒體功能

2025年9月10日,美國以及意大利學者聯(lián)合在Nature期刊上發(fā)表了題名為“ABCA7 variants impact phosphatidylcholine and mitochondria in neurons”的研究論文,揭示了ABCA7功能缺失變異通過破壞磷脂酰膽堿代謝介導阿爾茨海默病風險的機制。
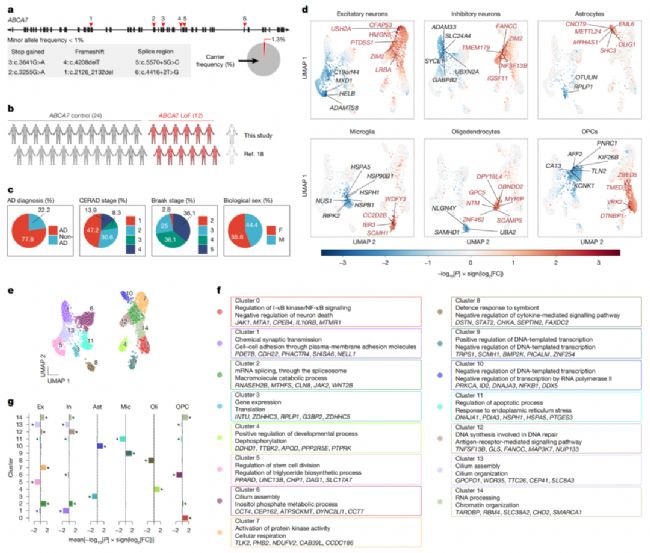
通過對人腦樣本的單核RNA測序分析,發(fā)現(xiàn)ABCA7功能缺失變異引起多種神經(jīng)細胞基因表達紊亂,其中高表達ABCA7的興奮性神經(jīng)元出現(xiàn)脂代謝、線粒體功能和DNA修復等通路異常。分子動力學模擬顯示常見致病突變ABCA7 p.Ala1527Gly同樣導致結構異常。iPS來源的神經(jīng)元模型證實ABCA7缺陷會導致線粒體功能障礙、氧化應激增加和磷脂酰膽堿代謝紊亂。補充CDP-膽堿可增強磷脂酰膽堿合成,逆轉(zhuǎn)上述異常,并改善β淀粉樣蛋白分泌和神經(jīng)元過度興奮這兩個阿爾茨海默病關鍵特征。
該研究不僅闡明ABCA7相關疾病風險機制,更為治療干預提供了新靶點。
DOI:10.1038/s41586-025-09520-y
8、調(diào)控持續(xù)性疼痛需求狀態(tài)的中樞樞紐是?

2025年10月8日,美國學者在Nature期刊上發(fā)表了題名為“A parabrachial hub for need-state control of enduring pain”的研究論文,揭示了臂旁核中表達Y1神經(jīng)肽受體的神經(jīng)元群體在持續(xù)性疼痛調(diào)控中的核心作用。
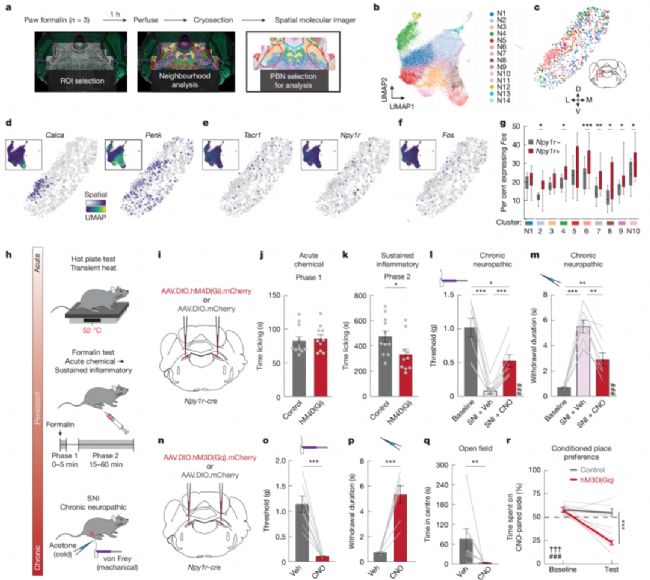
通過空間轉(zhuǎn)錄組學與神經(jīng)調(diào)控技術,發(fā)現(xiàn)組織損傷后臂旁核Y1R神經(jīng)元活性持續(xù)升高,且其活動水平可預測個體的功能代償行為。研究證實饑餓、口渴或天敵線索等生理需求能通過釋放神經(jīng)肽Y抑制Y1R神經(jīng)元活動,從而廣泛抑制不同類型損傷引發(fā)的持續(xù)性疼痛。這種需求狀態(tài)驅(qū)動的鎮(zhèn)痛效應不依賴于組織愈合狀態(tài),揭示了內(nèi)源性鎮(zhèn)痛系統(tǒng)通過特定神經(jīng)元集群實現(xiàn)多模態(tài)感覺整合的新機制。
該研究確立了臂旁核Y1R神經(jīng)元作為整合生理需求與疼痛信號的中樞樞紐,為理解慢性疼痛機制提供了新視角。
DOI:10.1038/s41586-025-09602-x

- LASER系列活體成像系統(tǒng)助力軟棗獼猴桃花青素調(diào)控機制解析
- 熱點速遞:近紅外納米探針合成技術顯著提升其近紅外發(fā)光強度與穩(wěn)定性
- 負載BMSC源性凋亡囊泡的纖維支架通過誘導巨噬細胞極化促進傷口愈合
- 具有半乳糖靶向的NIR-II納米探針實現(xiàn)原位肝癌手術切緣及深度精準成像
- 論文解讀:功能超聲成像(fUS)用于超早期卒中階段監(jiān)測的研究案例
- 文獻速遞:頂刊CNS神經(jīng)領域研究新進展11月(下)
- 利用1880-2080納米窗口進行高對比度活體熒光成像的新方法
- 供體平面性誘導分子扭曲構建D-A-D型AIE實現(xiàn)高效NIR-II熒光與光熱治療