ADC PK模型的特點、影響因素及建模方法
文章來源公眾號:ATCG 作者:adcg
劉瑜在《送你一顆子彈》里寫道:“一個人要像一支隊伍。”ADC也如此。
ADC的異質性使其擁有了一支攜帶不同DAR值的隊伍,在腫瘤治療前線沖鋒陷陣。
當然,這種異質性同時也給生物分析和PK建模方法帶來了挑戰。
一起來看看吧!
DOI鏈接:10.1208/s12248-020-00475-8
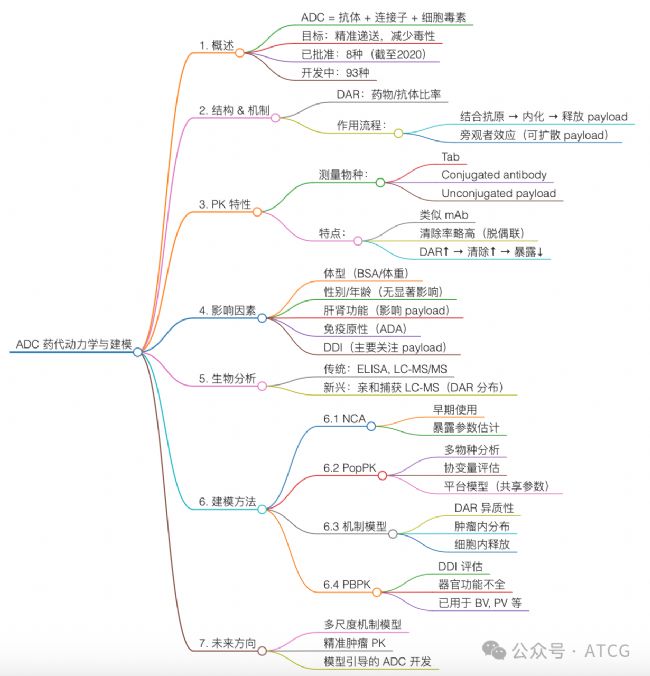
圖1 思維導圖
ADC PK的特點
ADC由三部分組成,因此它的PK具有裸抗和小分子的特性,并且以裸抗為主導。
由于和裸抗的分子量幾乎相同,ADC 在吸收、分布代謝和消除 (ADME) 方面與未偶聯的 mAb 具有相似的特征。例如具有與單抗類似的長半衰期(得益于FcRn介導的回收)、低清除率、低分布容積,以及潛在的靶介導藥物處置(dispostion)和非線性PK。但ADC的清除率通常高于其對應的裸抗,半衰期更短。主要原因是在蛋白水解降解途徑之外,增加了去偶聯這一額外的清除途徑。
并且,ADC在體內的PK是動態變化的。高DAR值的ADC species清除更快,導致平均DAR在血液循環中逐漸下降,同時系統內payload的水平上升。尤其是在不同患者體內,高度可變的單核吞噬細胞系統和其他免疫細胞上表達的FcγR會導致患者間的ADC異質性。以上產生的體內高度不一致的ADC濃度,與釋放payload和患者間腫瘤靶標大小的復雜過程的變異性結合,使得payload在體循環中PK的變異性更大。
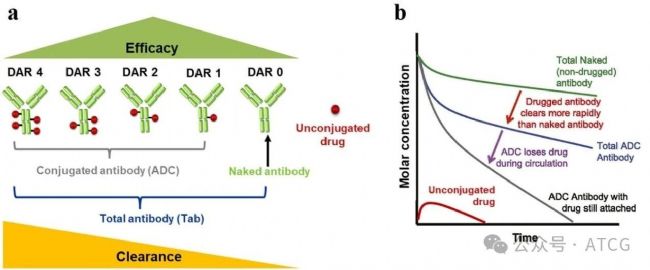
圖2 ADC的DAR分布和血液中清除率之間的關系
因此如圖2所示,ADC的PK特征總結如下:
| 成分 | PK特點 |
| Tab(總抗體) | - PK曲線最平穩,暴露量最高 - 是偶聯抗體的“母體”,兩者濃度高度相關 - 清除僅由抗體的消除驅動 |
| ADC | - 清除速度比Tab更快,受到抗體消除和載荷去偶聯的共同影響 - 通常被認為是與療效最直接相關的species |
| Payload | - 系統暴露量遠低于抗體類species - 因其釋放速率受ADC的PK影響,半衰期可能比其“天然”小分子形式更長, - 因其具有強細胞毒性,是安全毒性的主要關注點 |
影響PK的因素
01 結構設計
連接子穩定性:不穩定的連接子會導致在到達靶點前過早釋放payload,增加全身毒性,降低療效。
DAR值與載荷疏水性:高DAR和疏水性payload會增加ADC的聚集傾向和非特異性清除,導致PK表現不佳。
02 患者因素
體型:通常根據患者體表面積或體重給藥。
靶點表達與腫瘤負荷:影響靶點介導的清除,可能導致非線性PK。
肝腎功能:肝臟和腎臟是大多數小分子毒素代謝和排泄的主要器官,主要影響payload的暴露,對ADC抗體部分影響較小。
免疫原性:產生的抗藥抗體可能加速ADC的清除,降低其暴露量。
03 DDI(藥物相互作用)
已上市藥物顯示,ADC的抗體部分發生DDI的風險極低,因此主要關注payload帶來的DDI。
研究顯示brentuximab vedotin的游離載荷MMAE,和CYP450酶或轉運體的底物/抑制劑發生了DDI。
常規的分析方法有兩類,分別針對于大分子部分和小分子部分。針對于裸抗部分的是ELISA,針對于小分子部分為LC-MS/MS。近來有另一種方法——親和捕獲結合LC-MS/MS來給ADC中的DAR species定量。
以上的分析方法直接決定了PK模型的復雜程度和可靠性。
ADC的PK建模方法
ADC的PK建模經歷了從實證(empirical)為主的方法到基于分子機制的建模。以下是由簡單到復雜的過程:
01 Noncompartmental Analysis(NCA)
NCA是目前各國藥品監管機構(如FDA、NMPA)普遍接受和要求的標準分析方法(特別是在生物等效性研究中)。NCA對實測的血藥濃度-時間數據進行分析,通過數學計算得出描述藥物暴露情況的參數。
它的核心參數:
Cmax,曲線下面積(AUC),半衰期(t½),達到Cmax的時間(tmax),清除率(CL)和分布容積(Vd)等。
在早期研究(如首次人體試驗)中,樣本量小,首要任務是快速了解ADC及其載荷的基本暴露特征(如半衰期多長、暴露量多大),NCA是完全夠用的工具。當建立復雜的群體PK或PBPK模型后,可以用NCA計算的群體平均濃度來驗證模型預測是否合理。對于某些ADC,如果其游離載荷的濃度極低或與臨床結果無關,那么一個簡單的、基于偶聯ADC的NCA分析可能就足以支持劑量選擇,無需構建復雜模型。
02 Population PK modeling(PopPK)
PopPK是一種將所有受試者的數據同時納入一個模型,來估算PK參數的平均值(典型值)和變異性的方法。與NCA相比,PopPK建模方法可以同時使用所有患者在所有情況下的多種分析物的數據,以獲得固定(典型平均參數值)和隨機效應(受試者間和場間變異性)的無偏參數分布,從而產生更準確的總體估計。
它的核心參數是固定效應和隨機效應:
-
固定效應描述群體典型值的參數(如典型清除率)
-
隨機效應包括個體間變異(不同患者之間PK參數的天然差異),個體內變異(同一患者在不同時間點的測量波動),殘差變異(模型無法解釋的隨機誤差)和協變量效應(年齡、體重、肝腎功能等因素)。
PopPK用于分析臨床數據,回答諸如:“腎功能不全的患者是否需要調整劑量?”、“體重對ADC暴露量影響有多大?”等問題,從而優化整個群體的給藥方案。
03 Mechanism-Based Modeling
基于機制的PK模型是將藥物在體內發生的已知生物學、生理學過程,用數學方程明確表達出來的模型。它的目標不是簡單地擬合數據,而是解釋數據背后的“為什么”。已被用于支持藥物開發臨床前階段的候選藥物選擇,以及臨床I 期劑量選擇,從而將藥物作用從動物轉化為人類。
它的核心參數如下:
-
基于生物學過程的參數:如靶點結合速率、內化速率、細胞內載荷釋放速率、DAR依賴性清除率、腫瘤穿透率等。
圖5展示了一個異質性ADC的基于機制的PK模型。傳統的PK模型只用一個房室來描述“偶聯抗體”(一個平均值)。而這個機制模型(圖5b)為每一個DAR物種(DAR0到DAR8)都設置了一個獨立的“房室”。這些房室通過 “去偶聯” 過程連接:例如,一個DAR4的分子失去一個Payload,就變成了一個DAR3的分子,并進入DAR3的房室。它的關鍵機制是:DAR值越高,脫偶聯的速率越快(如圖中從DAR8到DAR7的速率常數 是最大的)。這是因為高DAR分子更不穩定、疏水性更強,更容易被清除。
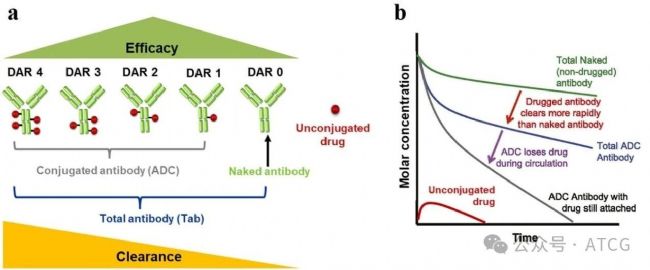
圖3 異質性ADC的基于機制的PK模型
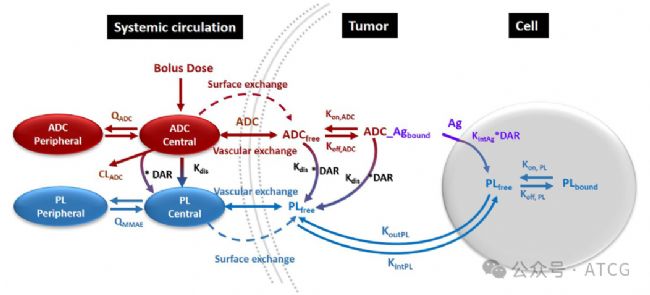
圖6展示了ADC與Payload的多尺度PK/PD建模。這個模型將整個過程分為幾個關鍵的“尺度”:系統,腫瘤組織,細胞內,并將細胞內Payload的濃度與腫瘤生長抑制或細胞殺傷效應通過藥效動力學模型聯系起來。該模型實現了從動物到人體的劑量預測,優化給藥方案,并擴展至“旁觀者效應”建模。
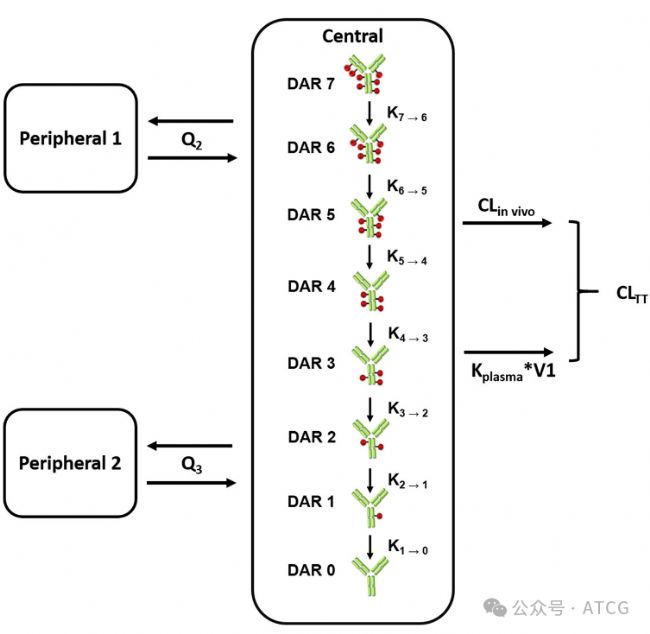
圖4 ADC和有效載荷PK在體循環中的多尺度建模示意圖,并在與細胞表面的抗原 (Ag) 結合后分布到腫瘤組織和細胞內區域
04 Physiologically- Based Modeling (PBPK)
PBPK是機制性模型的一個特殊和更具體的子類。它通過構建一個基于真實人體生理結構的模型來預測藥物在全身各器官和組織中的處置(disposition)過程。PBPK具有極強的機制性和預測能力,特別適合評估肝/腎功能不全和藥物相互作用,因為可以直接修改模型中的肝臟大小或酶活性來模擬這些情況。
它的核心參數如下:
-
生理參數:器官體積、組織血流速率、酶表達水平等(通常來自文獻)。
-
藥物特異性參數:藥物的脂溶性、分子大小、與組織和血漿蛋白的結合率、酶促反應參數等。
PBPK在評估DDI風險方面具有很大作用。已用在brentuximab vedotin與咪達唑侖(CYP3A 底物)、酮康唑(P-gp 和強 CYP3A 抑制劑)和利福平(P-gp 和強 CYP3A 誘導劑)的DDI評估上,并得出與臨床 DDI 研究觀察結果相當的預測結果。并且,POLIVY®的DDI評估就是完全基于PBPK模型獲批的,替代或補充復雜的臨床DDI研究。
總結(AI打分)| PK模型 | 常用度 | 潛力 | 主要應用場景 |
| NCA | 5 | 1 | 早期臨床階段的基礎PK參數估算;監管報批的標準方法 |
| PopPK | 4 | 2 | 臨床開發階段,量化變異、優化劑量、評估協變量(如腎功能)影響 |
| 機制模型 | 2 | 5 | 臨床前至臨床轉化;候選藥物優化;理解藥物作用機制;預測腫瘤內濃度。 |
| PBPK | 1 | 4 | 評估藥物相互作用和器官功能不全的影響;支持監管決策 |
一些思考
- PK模型越復雜越好嗎?還是夠用就行?
- 分析方法產生的數據如何影響建模?