
文獻解讀:內(nèi)皮細胞中GTPBP3在血管生成中的作用
GTP結(jié)合蛋白3(GTPBP3)是一種高度保守的tRNA修飾酶,是5-牛磺酸甲基尿苷(τm5U)生物合成的關(guān)鍵酶,與細胞內(nèi)線粒體功能障礙有關(guān),但其在不同細胞類型血管發(fā)育和血管生成中的具體作用尚不清楚。2025年6月,中南大學湘雅二醫(yī)院于碧蓮教授研究團隊在Angiogenesis (IF 9.2)期刊發(fā)表題為“Endothelial GTPBP3 directs developmental angiogenesis and neovascularization after limb ischemia via the mtROS/HRl/ATF4/mTORC1 axis”的文章。本研究探討了內(nèi)皮細胞中GTPBP3在血管生成中的作用,發(fā)現(xiàn)其通過mtROS/HRl/ATF4/mTORC1軸調(diào)控發(fā)育性血管生成和肢體缺血后的新血管形成,凸顯了GTPBP3作為血管生成相關(guān)疾病潛在治療靶點的價值。
· 維真助力·
基因信息:GTPBP3,GTP結(jié)合蛋白3
病毒產(chǎn)品:Ad-GTPBP3、Ad-con
感染細胞:HUVECs
MOI:100
檢測時間:感染后48h
· 維真助力·
基因信息:GTPBP3,GTP結(jié)合蛋白3
病毒產(chǎn)品:Ad-GTPBP3、Ad-con
感染細胞:HUVECs
MOI:100
檢測時間:感染后48h

腺病毒有效介導GTPBP3在HUVECs中過表達
研究結(jié)果
1、GTPBP3在內(nèi)皮細胞血管生成中起關(guān)鍵作用
研究數(shù)據(jù)顯示血管生成刺激增加內(nèi)皮細胞GTPBP3的表達水平。在內(nèi)皮細胞特異性GTPBP3敲除小鼠中,胚胎致死率高,胚胎發(fā)育不良且血管網(wǎng)絡存在缺陷,視網(wǎng)膜血管的總血管長度和分支點減少;而髓系細胞特異性敲除GTPBP3的小鼠無胚胎致死現(xiàn)象,表明內(nèi)皮細胞中GTPBP3對胚胎血管發(fā)育至關(guān)重要。研究團隊進一步構(gòu)建了通過他莫昔芬誘導的內(nèi)皮細胞特異性GTPBP3敲除小鼠模型,發(fā)現(xiàn)其出生后視網(wǎng)膜血管徑向擴展延遲,血管密度、總血管長度、分支點及絲狀偽足數(shù)量和長度均減少,增殖內(nèi)皮細胞數(shù)量降低;在成年小鼠后肢缺血模型中,該敲除小鼠血流恢復緩慢,再生肌區(qū)的毛細血管(CD31)和小動脈(α-SMA)密度顯著降低,表明內(nèi)皮細胞GTPBP3對出生后視網(wǎng)膜血管生成和肢體缺血后的新血管形成至關(guān)重要。體外實驗中,GTPBP3敲低抑制HUVECs管形成、增殖和遷移,而腺病毒載體介導的過表達則促進這些過程。以上數(shù)據(jù)表明GTPBP3對內(nèi)皮細胞的血管生成活性至關(guān)重要。
1、GTPBP3在內(nèi)皮細胞血管生成中起關(guān)鍵作用
研究數(shù)據(jù)顯示血管生成刺激增加內(nèi)皮細胞GTPBP3的表達水平。在內(nèi)皮細胞特異性GTPBP3敲除小鼠中,胚胎致死率高,胚胎發(fā)育不良且血管網(wǎng)絡存在缺陷,視網(wǎng)膜血管的總血管長度和分支點減少;而髓系細胞特異性敲除GTPBP3的小鼠無胚胎致死現(xiàn)象,表明內(nèi)皮細胞中GTPBP3對胚胎血管發(fā)育至關(guān)重要。研究團隊進一步構(gòu)建了通過他莫昔芬誘導的內(nèi)皮細胞特異性GTPBP3敲除小鼠模型,發(fā)現(xiàn)其出生后視網(wǎng)膜血管徑向擴展延遲,血管密度、總血管長度、分支點及絲狀偽足數(shù)量和長度均減少,增殖內(nèi)皮細胞數(shù)量降低;在成年小鼠后肢缺血模型中,該敲除小鼠血流恢復緩慢,再生肌區(qū)的毛細血管(CD31)和小動脈(α-SMA)密度顯著降低,表明內(nèi)皮細胞GTPBP3對出生后視網(wǎng)膜血管生成和肢體缺血后的新血管形成至關(guān)重要。體外實驗中,GTPBP3敲低抑制HUVECs管形成、增殖和遷移,而腺病毒載體介導的過表達則促進這些過程。以上數(shù)據(jù)表明GTPBP3對內(nèi)皮細胞的血管生成活性至關(guān)重要。
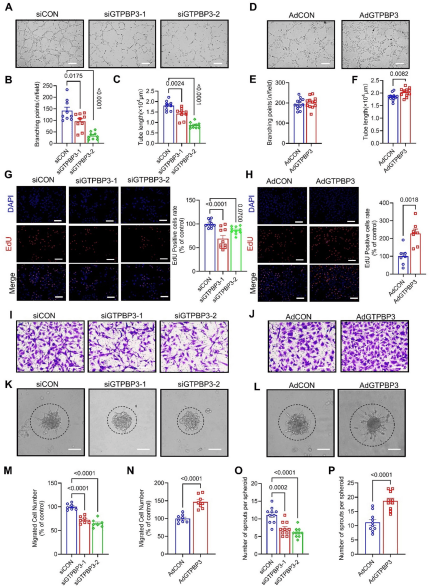
GTPBP3是體外內(nèi)皮血管生成活性所必需的
2、GTPBP3通過激活mTORC1信號通路促進血管生成
血管內(nèi)皮生長因子(VEGF)信號傳導和哺乳動物雷帕霉素靶蛋白(mTOR)通路是血管生成的關(guān)鍵調(diào)節(jié)因子。但在GTPBP3敲低或過表達后,VEGFA和VEGF受體2 mRNA表達水平保持不變,此外,用VEGF信號調(diào)節(jié)因子的抑制劑處理細胞,進一步排除了VEGF信號傳導作為GTPBP3驅(qū)動的血管生成的介導物。對mTORC1途徑的研究表明,GTPBP3的過表達顯著增加了HUVEC中mTORC1的活化,GTPBP3敲低得到了相反的結(jié)果;并且GTPBP3相關(guān)的mTORC1激活獨立于AKT信號傳導發(fā)生。使用mTORC1復合物的有效激活劑亮氨酸的拯救實驗進一步驗證了mTORC1通路參與GTPBP3誘導的血管生成。通過研究GTPBP3抑制如何影響mTORC1活性,證實AMPK和HRI/ATF4/Sestrin2通路響應GTPBP3的改變,抑制HRI/ATF4/Sestrin 2通路可挽救缺陷型GTPBP3缺陷誘導的血管生成。
血管內(nèi)皮生長因子(VEGF)信號傳導和哺乳動物雷帕霉素靶蛋白(mTOR)通路是血管生成的關(guān)鍵調(diào)節(jié)因子。但在GTPBP3敲低或過表達后,VEGFA和VEGF受體2 mRNA表達水平保持不變,此外,用VEGF信號調(diào)節(jié)因子的抑制劑處理細胞,進一步排除了VEGF信號傳導作為GTPBP3驅(qū)動的血管生成的介導物。對mTORC1途徑的研究表明,GTPBP3的過表達顯著增加了HUVEC中mTORC1的活化,GTPBP3敲低得到了相反的結(jié)果;并且GTPBP3相關(guān)的mTORC1激活獨立于AKT信號傳導發(fā)生。使用mTORC1復合物的有效激活劑亮氨酸的拯救實驗進一步驗證了mTORC1通路參與GTPBP3誘導的血管生成。通過研究GTPBP3抑制如何影響mTORC1活性,證實AMPK和HRI/ATF4/Sestrin2通路響應GTPBP3的改變,抑制HRI/ATF4/Sestrin 2通路可挽救缺陷型GTPBP3缺陷誘導的血管生成。
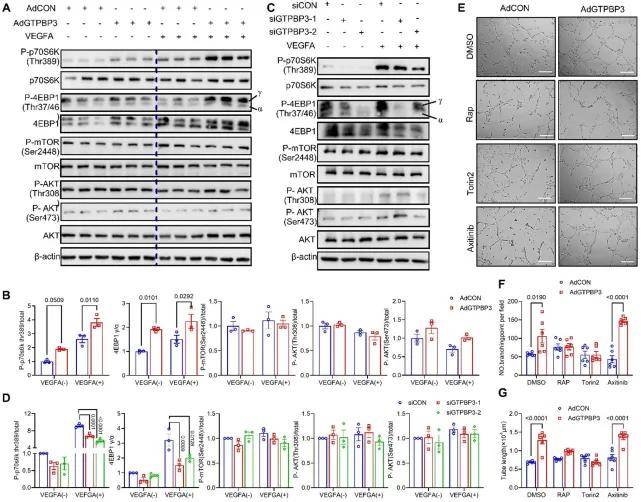
GTPBP3通過激活mTORC1信號通路調(diào)節(jié)血管生成
3、內(nèi)皮細胞GTPBP3缺陷誘導線粒體功能障礙
研究表明線粒體應激可激活整合應激反應(ISR)途徑中的HRI分支。為研究GTPBP3缺陷型HUVEC中的HRI活化機制,研究團隊評估了GTPBP3在EC內(nèi)線粒體功能中的作用。在HUVECs中敲低GTPBP3降低了線粒體膜電位及氧消耗率,雖ATP水平無顯著變化,但AMP/ATP比值升高、NAD+/NADH比值降低,且線粒體活性氧積累;同時,GTPBP3缺失會激活HRI/eIF2α/ATF4通路,使磷酸化eIF2α、ATF4及Sestrin2水平上調(diào),而線粒體ROS清除劑MitoQ可逆轉(zhuǎn)這些變化,表明GTPBP3缺失誘導的線粒體功能異常會導致mtROS積累,進而激活HRI/eIF2α/ATF4通路。進一步分析證明靶向mtROS可挽救GTPBP3缺陷引起的體內(nèi)外血管生成障礙。
研究表明線粒體應激可激活整合應激反應(ISR)途徑中的HRI分支。為研究GTPBP3缺陷型HUVEC中的HRI活化機制,研究團隊評估了GTPBP3在EC內(nèi)線粒體功能中的作用。在HUVECs中敲低GTPBP3降低了線粒體膜電位及氧消耗率,雖ATP水平無顯著變化,但AMP/ATP比值升高、NAD+/NADH比值降低,且線粒體活性氧積累;同時,GTPBP3缺失會激活HRI/eIF2α/ATF4通路,使磷酸化eIF2α、ATF4及Sestrin2水平上調(diào),而線粒體ROS清除劑MitoQ可逆轉(zhuǎn)這些變化,表明GTPBP3缺失誘導的線粒體功能異常會導致mtROS積累,進而激活HRI/eIF2α/ATF4通路。進一步分析證明靶向mtROS可挽救GTPBP3缺陷引起的體內(nèi)外血管生成障礙。
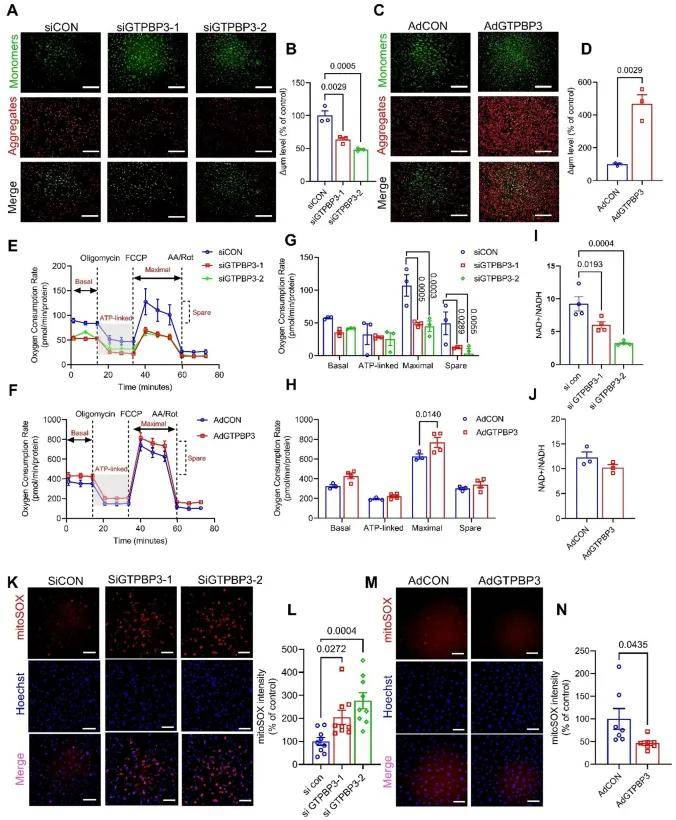
內(nèi)皮細胞GTPBP3的缺失損害EC線粒體功能
結(jié)論
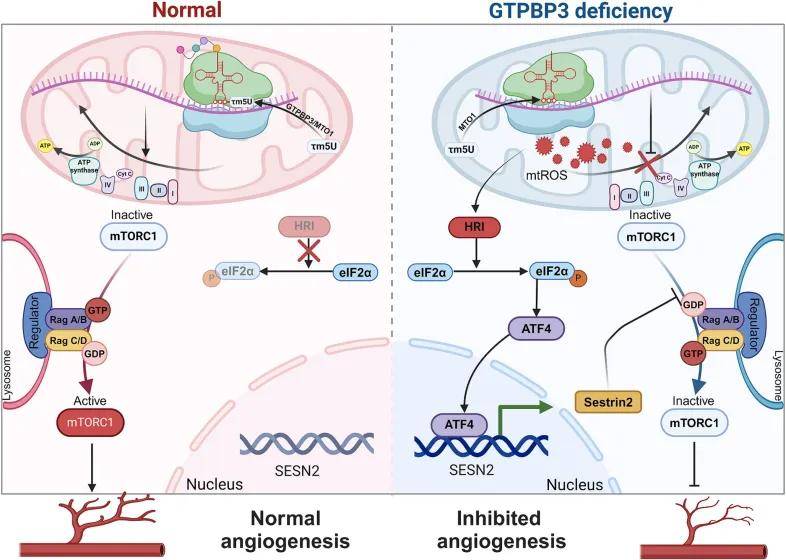
本研究闡明了GTPBP3在EC功能和血管生成中的關(guān)鍵作用。GTPBP3缺陷通過獨立于VEGF信號的mTORC1信號通路導致血管生成受損。血管生成缺陷與mtROS誘導的HRI/ATF4/Sestrin2信號通路的激活有關(guān)。這些發(fā)現(xiàn)強調(diào)了線粒體功能在內(nèi)皮細胞中的重要性,為血管生成相關(guān)疾病的潛在治療靶點提供了新的見解。
本研究闡明了GTPBP3在EC功能和血管生成中的關(guān)鍵作用。GTPBP3缺陷通過獨立于VEGF信號的mTORC1信號通路導致血管生成受損。血管生成缺陷與mtROS誘導的HRI/ATF4/Sestrin2信號通路的激活有關(guān)。這些發(fā)現(xiàn)強調(diào)了線粒體功能在內(nèi)皮細胞中的重要性,為血管生成相關(guān)疾病的潛在治療靶點提供了新的見解。

- 增強AAV對腎臟組織的感染效率的方法分享
- 文獻:AAA中內(nèi)皮細胞在單細胞水平上的異質(zhì)性和轉(zhuǎn)錄特征
- 鄰位連接技術(shù)(PLA)的原理、實驗流程及在生命科學研究中的應用
- 衰老標志的因果關(guān)系:端粒/rDNA才是根本因素?
- 蛋白質(zhì)結(jié)構(gòu)預測的技術(shù)變革、新挑戰(zhàn)與發(fā)展方向
- 空間多組學助力揭示膠質(zhì)瘤中三級淋巴結(jié)構(gòu)的臨床意義
- CZ CELLxGENE Discover生物數(shù)據(jù)庫使用步驟介紹
- CosMx SMI空間原位技術(shù)助力無需組織解離實現(xiàn)單細胞的多靶標檢測



Copyright(C) 1998-2025 生物器材網(wǎng) 電話:021-64166852;13621656896 E-mail:info@bio-equip.com