多組學分析揭示DNA低甲基化定義人組織調節性T細胞的表觀遺傳適應性
2025年7月16日,德國雷根斯堡大學Markus Feuerer教授和美因茨大學Michael Delacher 教授團隊合作,通過整合人類皮膚組織和血液中調節性T細胞(Regulatory T cells,Treg細胞)的全基因組亞硫酸鹽測序(WGBS)、染色質可及性測序(ATAC-seq)和轉錄組測序(RNA-seq)多組學數據,系統解析了人類皮膚組織和血液中Treg細胞的表觀遺傳特征。本研究發現皮膚Treg細胞表現出以DNA低甲基化為主的組織適應性呈現狀態,尤其發生在轉座子元件(TE)區域。進一步研究證實,血液中CCR8+ Treg細胞在DNA甲基化模式上與皮膚Treg高度相似,但在染色質可及性和基因表達上部分差異,提示其可能是從皮膚組織再循環至血液的Treg細胞。該研究首次利用多組學定義了人多組織Treg細胞的表觀遺傳適應性狀態,并提出了CCR8+ Treg作為循環中組織來源Treg的甲基化標志。相關研究成果以“DNA hypomethylation traits define human regulatory T cells in cutaneous tissue and identify their blood recirculating counterparts”為題發表在免疫學相關領域頂刊《Nature immunology》。

英文標題:DNA hypomethylation traits define human regulatory T cells in cutaneous tissue and identify their blood recirculating counterparts
中文標題:DNA低甲基化表征定義了皮膚組織中的人類調節性T細胞,并鑒定出其血液再循環對應物
發表時間:2025年7月16日
發表期刊:Nature immunology
影響因子:IF27.6/Q1
技術平臺:WGBS、ATAC-seq、RNA-seq
作者單位:雷根斯堡大學、美因茨大學等
DOI:10.1038/s41590-025-02210-x
組織中廣泛存在的CD4+ Treg細胞發揮著至關重要的免疫調節和再生作用,但其表觀遺傳特征和分化機制仍未闡明。本研究對人的皮膚和血液Treg細胞進行全基因組DNA甲基化分析,并將其與染色質可及性和基因表達進行多組學關聯分析,鑒定出調控皮膚Treg細胞的組織適應性程序。此外,研究發現轉座元件亞家族是組織中Treg細胞低甲基化圖譜的主要組分。有趣的是,基于T細胞抗原受體序列和DNA低甲基化同源性,本研究分析表明,血液CCR8+ Treg細胞中可能含有再循環的人類皮膚Treg細胞,而染色質可及性和基因表達差異分析表明,其組織適應程序在再循環過程中發生了一定程度的逆轉。本研究結果為深入理解人類組織Treg細胞的生物學特性提供了見解,也為Treg細胞靶向治療的安全性評估提供了重要的理論依據。
研究方法
樣本來源:
- 皮膚組織Treg細胞:來自7名健康女性捐贈者的皮膚組織。
- 血液Treg細胞:從10例健康女性血小板捐贈者的白細胞減除腔中分離外周血單核細胞(PBMCs),包括CCR8+ Treg、CD45RA+ naive Treg和常規T細胞(Tconv)。
- 脂肪組織Treg細胞:來自皮下脂肪,用于驗證組織保守性。
表觀多組學測序分析:
- WGBS(全基因組亞硫酸鹽測序):用于全基因組DNA甲基化分析,覆蓋約2.8M CpG位點,中位覆蓋度2–7×。
- ATAC-seq(染色質可及性測序):利用公開數據及自行生成的數據,分析染色質開放區域。
- RNA-seq(轉錄組測序):分析基因表達差異,部分數據來源于公開數據庫。
- 多組學整合:通過重疊DMRs、差異可及性峰值和差異表達基因,構建“DMR–peak–gene links”關聯調控網絡。
驗證分析:
- T細胞受體(TCR)追蹤:利用scRNA-seq/scTCR-seq數據,通過TCR克隆型重疊評估細胞來源同一性。
- 小鼠模型驗證:使用光轉換Kaede小鼠(Tg(CAG-Kaede)15Kgwa)追蹤皮膚Treg細胞向引流淋巴結的遷移,并結合CCR8表達分析。
(1)DNA低甲基化定義皮膚和血液CCR8+ Treg細胞
本研究首先通過WGBS技術對來自9例健康女性供者的血液naïve Treg細胞(CD45RA+CCR8-)、血液naïve Tconv細胞、血液CCR8+Treg細胞(CD45RA-CCR8+)、皮膚Treg細胞和皮膚Tconv細胞共5種CD4+ T細胞亞群的全基因組DNA甲基化進行深度測序分析。分析結果顯示,皮膚Treg細胞和血液CCR8+ Treg細胞的全基因組DNA甲基化水平上表現出顯著的DNA低甲基化表征,顯著區別于血液CD45RA+ Treg/Tconv細胞,表明這兩類細胞核心表觀遺傳程序共性。為精確定義細胞類型特異性甲基化特征,研究團隊開發"signature region"算法用于篩選甲基化差異≥0.15、連續至少3個CpG、間隔<300 bp的區域。該算法共鑒定出182,204個特異性標記區域,低甲基化特征同時存在于皮膚Treg、皮膚Tconv和血液CCR8+ Treg細胞中,且這些區域主要富集于基因間區,遠離CpG島,表明低甲基化可能主要影響非編碼調控元件。該結果首次從全基因組甲基化層面證實血液CCR8+ Treg細胞與組織Treg細胞的表觀遺傳共性。
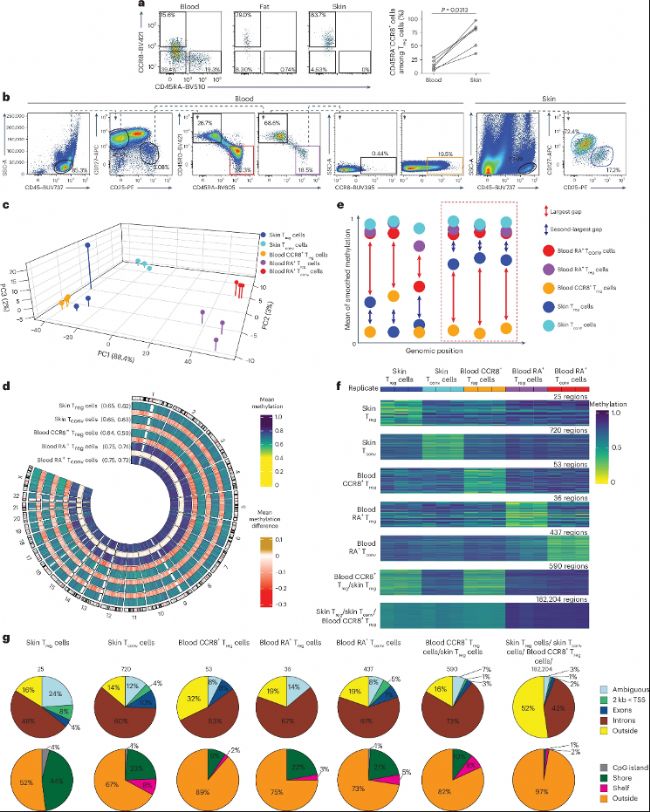
圖1:皮膚Treg細胞和血液CCR8+ Treg細胞與血液CD45RA+ Treg細胞的DNA甲基化分析
a. 流式細胞術代表性圖譜顯示人類血液、脂肪和皮膚中CD4+CD127-CD25+ Treg細胞亞群的CD45RA-CCR8+ Treg細胞(左圖),以及人類血液與皮膚中CD4+ CD127-CD25+ Treg細胞亞群的CD45RA-CCR8+ Treg細胞比例。
b. 分選策略示意圖顯示從1例健康女性供者中分離血液CD45RA+ Treg細胞、血液CD45RA+ Tconv細胞、血液CCR8+ Treg細胞、皮膚Tconv細胞和皮膚Treg細胞。
c. 對來自9例健康女性供者的血液和皮膚和血液中分離的血液CD45RA+ Treg細胞(RA+ Treg細胞)、血液CD45RA+ Tconv細胞(RA+ Tconv細胞)、血液CCR8+ Treg細胞、皮膚Tconv細胞和皮膚Treg細胞進行DNA甲基化主成分分析。
d. c中供者的人類血液CD45RA+ Treg細胞、血液CCR8+ Treg細胞、皮膚Tconv細胞和皮膚Treg細胞中按基因組位置顯示的甲基化水平,以及與血液CD45RA+ Tconv細胞的差異分析。
e. 任意兩種細胞類型之間最顯著甲基化差異(紅色箭頭)提取細胞類型特征,從而篩選出特征區域。
f. e中所選細胞類型特征區域的DNA甲基化情況。
g. e中特征區域的細胞類型特征在由基因定義的基因組區間(上圖)和CpG島(下圖)中的分布。
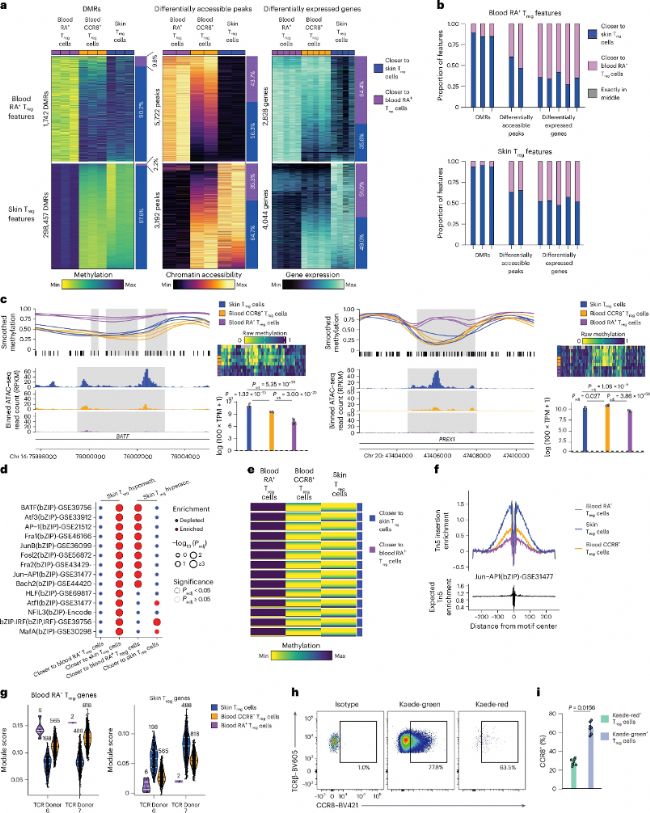
(2)多組學揭示差異甲基化區域-染色質開放峰-基因表達關聯調控網絡(DMR–peak–gene links)
為建立DNA甲基化與功能調控的直接聯系,研究整合WGBS數據與已發表的scATAC-seq及bulk RNA-seq數據,構建了"DMR-peak-gene"關聯分析調控網絡,即一個基因組區域同時存在差異甲基化、差異可及性和關聯基因差異表達。在比較血液CD45RA+ naïve Treg 和Tconv細胞,共鑒定出超3600個差異甲基化區域(DMRs),其中包含TNFRSF1B、TNFRSF9、IKZF2、IKZF4、FOXP3等經典Treg功能基因。進一步整合形成了151個DMR–peak–gene links,涉及73個基因。
在Treg表征位點(如FOXP3、TIGIT、IL2RA、CTLA4、TNFRSF1B)上,WGBS揭示的低甲基化模式與ATAC-seq檢測的染色質開放及RNA-seq檢測的高表達呈現顯著負相關。此外,研究通過靶基因亞硫酸鹽測序技術,在6例獨立男性供者中驗證了FOXP3、TIGIT、IL2RA、TNFRSF1B、LRRN3和SYTL3等位點的甲基化差異。這些關聯分析揭示了甲基化變化如何通過染色質可及性調控基因表達,為理解Treg細胞功能調控提供了多組學證據鏈。
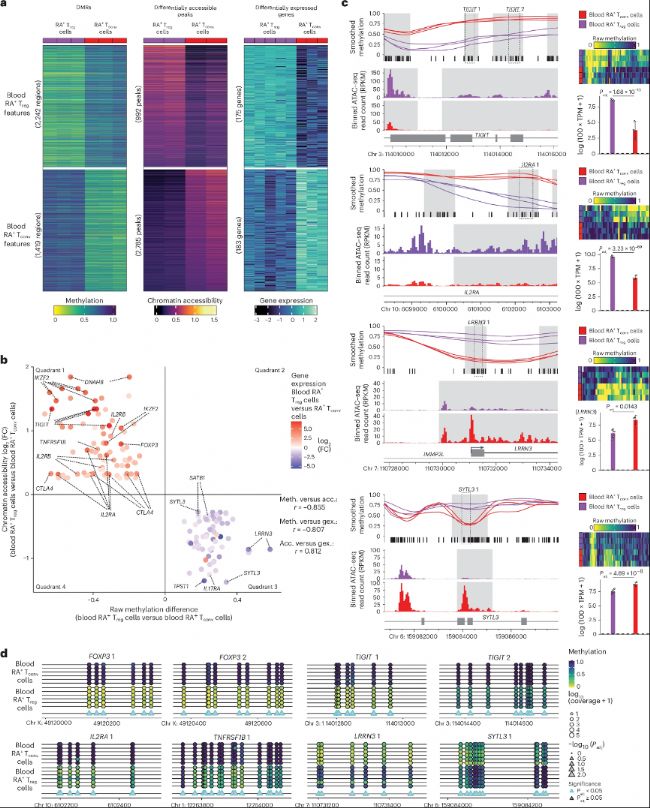
a. 血液CD45RA+ Treg細胞(RA+ Treg細胞)與血液CD45RA+ Tconv細胞(RA+ Tconv細胞)之間差異甲基化區域(左)、差異可及性peaks(中)和差異表達基因(右)的甲基化、染色質可及性和表達。
b. DMR–peak–gene關聯分析(n=151)中差異甲基化、可及性和表達之間的相關性。
c. 血液CD45RA+ Treg細胞和血液CD45RA+ Tconv細胞中選定DMR–peak–gene關聯的平滑甲基化(左上)、原始甲基化(右上)、染色質可及性(左下)和表達(右下)。
d. Taregt-BS測序顯示選定區域血液CD45RA+ Treg細胞與血液CD45RA+ Tconv細胞的甲基化差異。
(3)皮膚Treg細胞組織適應具有多組學特征
為研究皮膚Treg細胞的組織特異性適應過程,研究團隊鑒定出300,199個DMRs(其中298,457個為低甲基化)、8,914個差異可及性峰(3,192個高開放)和6,877個差異表達基因(4,049個高表達)。通過DMR-peak-gene關聯分析,發現1,203個三聯關聯中,813個表現為低甲基化-高開放-高表達模式,涉及TNFRSF8(CD30)、RELB、CCR8、PRDM1和BATF等關鍵轉錄因子。WGBS數據顯示,這些DMR甲基化水平變化程度遠高于染色質可及性變化,提示DNA甲基化可能是促進組織適應的"主開關"。功能富集分析表明,低甲基化關聯基因顯著富集于IL-2-STAT5信號、TNF信號、TGFβ信號等Treg細胞核心通路。值得注意的是,將皮膚Treg特征與脂肪組織Treg數據交叉驗證,發現脂肪Treg細胞中共有絕大多數低甲基化特征,證明了WGBS鑒定的表觀遺傳適應程序在不同組織的Treg細胞間具有保守性。該結果揭示,組織Treg細胞的適應過程本質上是一個以DNA去甲基化為主導的大規模表觀遺傳重編程事件,而非簡單的轉錄因子誘導表達。
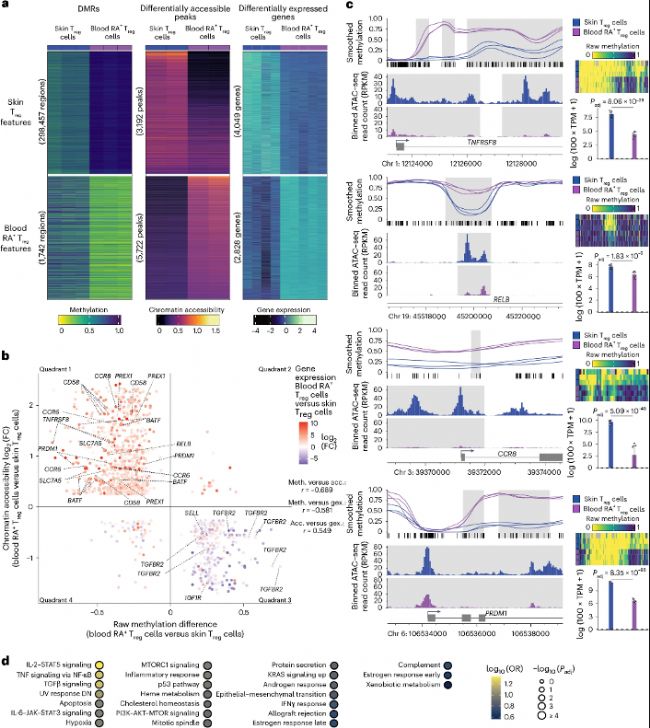
圖3:DMR–peak–gene關聯分析定義皮膚Treg細胞發育的多組學表征。
a. 皮膚Treg細胞與血液CD45RA+ Treg細胞(血液RA+ Treg細胞)之間差異甲基化區域(左)、差異可及性峰(中)和差異表達基因(右)的甲基化、染色質可及性和表達情況。
b. DMR–peak–gene關聯(n=1,203個)中差異甲基化、可及性和基因表達之間的相關性。
c. 所選DMR–peak–gene關聯分析顯示在皮膚Treg細胞和血液CD45RA+ Treg細胞中的平滑甲基化(左上)、原始甲基化(右上)、染色質可及性(左下)和基因表達(右下)。
d. 多組學分析顯示皮膚Treg細胞特征中基因的標記基因集富集情況。
(4)bZIP和bHLH motif的甲基化模式標記皮膚Treg細胞
通過解析DMRs中的轉錄因子結合位點,研究發現在皮膚Treg細胞低甲基化DMRs和高可及性峰中,bZIP轉錄因子(如BATF、Jun-AP1)的結合motifs中顯著富集。而bHLH轉錄因子(如c-Myc、n-Myc、USF1)motif僅富集于低甲基化DMRs,未在可及性峰值中富集。這些結果表明,bZIP因子可能在皮膚Treg細胞適應中同時受甲基化與可及性調控,而bHLH因子可能受甲基化調控進而發揮功能。
為驗證這些低甲基化位點功能,研究選取MLPH和GNA11位點的c-Myc結合位點,運用CRISPR激活系統(CRISPRa)進行靶向激活,結果顯示這兩個基因表達顯著上調,驗證了c-Myc結合位點低甲基化與鄰近基因(如MLPH、GNAI1)表達上調的相關性。這一功能實驗直接證明WGBS鑒定的低甲基化區域具有增強子活性,且其調控作用依賴于精確的甲基化狀態。
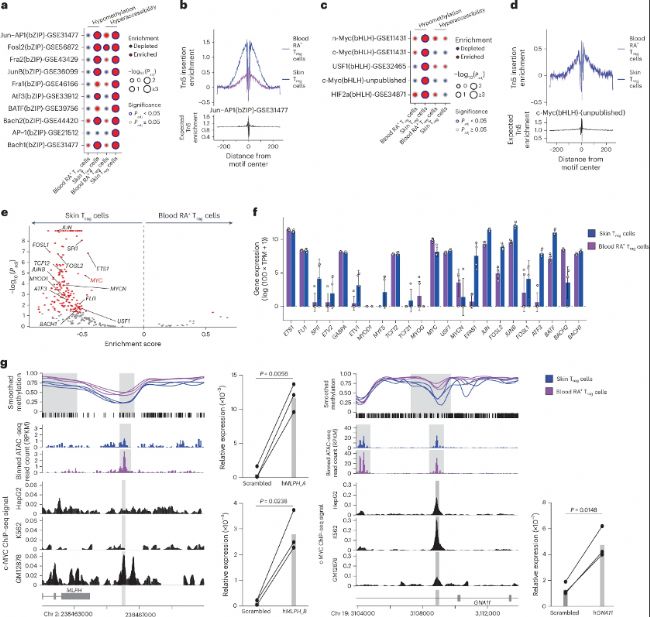
(5)血液CCR8+ Treg細胞與皮膚Treg細胞的DNA甲基化模式相似
研究將血液CCR8+ Treg細胞與皮膚Treg與血液naïve Treg的甲基化譜進行重疊分析,發現在97%的DMRs中,CCR8+ Treg細胞的甲基化水平更接近皮膚Treg細胞。然而,在染色質可及性(約60%)和基因表達(約45%)譜中,相似性顯著降低。這種"甲基化-可及性-表達"的梯度遞減現象提示,再循環Treg細胞在離開組織后,血液CCR8+ Treg細胞可能保留了組織Treg的甲基化“記憶”,但在循環過程中的部分可及性與表達程序被逆轉。
對BATF、PREX1和SPRED2等位點的進一步分析顯示,盡管這些區域的甲基化狀態在CCR8+ Treg中維持低水平,但染色質開放狀態未維持,已趨近初始Treg,揭示甲基化作為更穩定表觀遺傳標記的特性。小鼠Kaede光轉化實驗進一步證實,65%的遷出皮膚引流淋巴結的Treg細胞表達CCR8,而原位Treg僅30%表達,從體內動態角度驗證了CCR8+ Treg細胞的組織來源。該結果為確定血液CCR8+ Treg細胞作為組織駐留Treg細胞的循環對應物提供了證據。
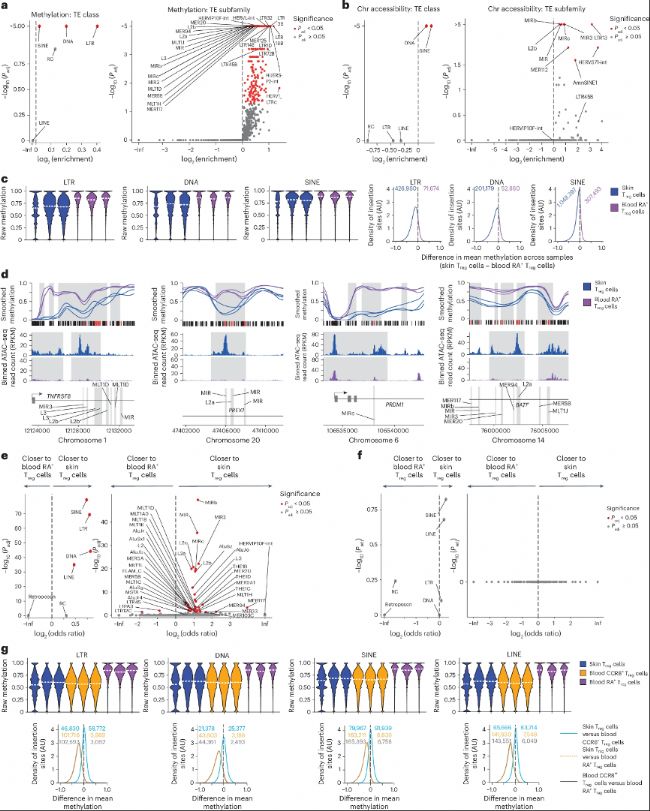
(6)轉座元件(TF)低甲基化是組織Treg細胞的標記
轉座子元件(TE)在人類基因組中占比約40–50%,常被DNA甲基化沉默。本研究發現,在皮膚Treg細胞中,SINE、LTR、DNA等TE亞家族(如LTR18B、HUERS-P2-int、MIR、L2a)在低甲基化DMRs中顯著富集。直接分析TE插入位點水平顯示,皮膚Treg細胞中LTR、SINE、DNA類TE的甲基化水平顯著低于血液CD45RA+ Treg。這些TE插入位點常位于TNFRS8、PREX1、PRDM1、BATF等皮膚Treg特征基因內部或鄰近區域,提示TE低甲基化可能通過調控鄰近基因表達參與組織適應性。
將血液CCR8+ Treg細胞納入比較后,發現這些細胞同樣富集TE低甲基化特征,且程度與皮膚Treg類似,但未在可及性中富集,進一步支持TE低甲基化是組織Treg細胞的穩定表觀遺傳印記。
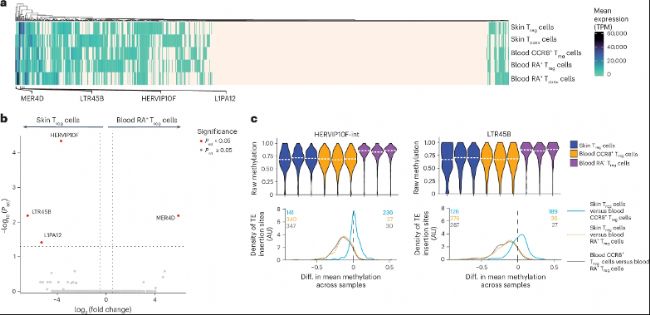
(7)TE亞家族在皮膚Treg細胞中獲得RNA表達
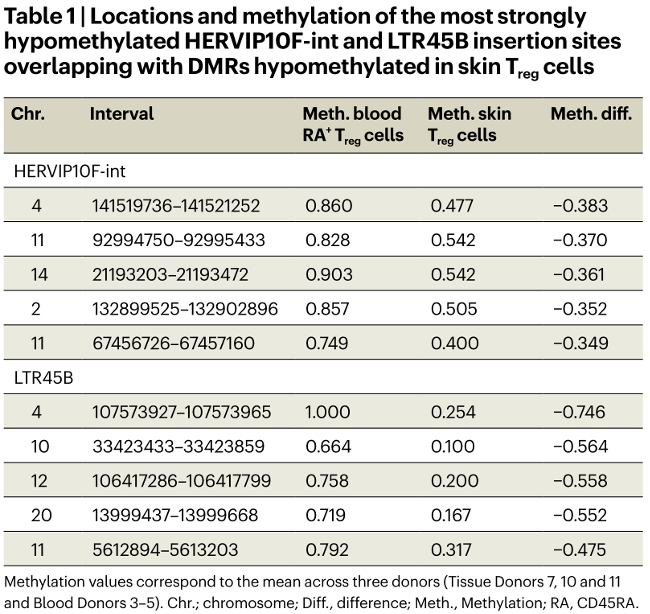
為驗證TE低甲基化是否導致其轉錄激活,研究進一步發現,部分TE亞家族在皮膚Treg細胞中轉錄上調,HERVIP10F-int和LTR45B在皮膚Treg細胞中顯著高表達。甲基化數據顯示,這兩個TE亞家族的插入位點在皮膚和脂肪Treg細胞中均呈現顯著低甲基化,且在血液CCR8+ Treg細胞中同樣保守。表1列出了5個最顯著低甲基化的HERVIP10F-int和LTR45B插入位點,其中,染色體4上的一個HERVIP10F-int插入位點在皮膚Treg中甲基化降低達74%。這表明TE不僅是低甲基化的被動載體,還可能通過轉錄活性參與Treg細胞的組織適應調控。該結果不僅為TE參與免疫調控提供了人類原代細胞證據,也暗示TE再激活可能是組織Treg細胞獲得組織修復功能的分子基礎之一。
結論和啟示
本研究建立了人類多組織Treg細胞高分辨率DNA甲基化圖譜,揭示以全基因組低甲基化和TE再激活為核心的組織適應性程序,并多層次證實血液CCR8+ Treg細胞是組織駐留Treg的再循環對應物。主要結論包括:
(1)DNA甲基化是定義組織Treg細胞身份穩定表觀遺傳標記;
(2)TE低甲基化是組織Treg特征的重要組分;
(3)血液CCR8+ Treg細胞保留組織甲基化記憶,但轉錄和染色質狀態部分恢復初始樣特征;
(4)靶向CCR8+ Treg細胞的治療需謹慎評估對組織穩態的影響。
WGBS作為本研究中全基因組DNA甲基化圖譜的核心關鍵技術:1.鑒定出組織與血液Treg細胞的甲基化差異;2.發現TE區域的廣泛低甲基化;3.通過甲基化相似性推斷細胞起源與遷移關系;4.整合多組學數據建立DMR–peak–gene調控網絡。該技術在此類研究中的優勢在于其全面性與分辨率,未來可廣泛適用于追溯細胞發育與遷移軌跡、解析復雜組織中細胞亞型的表觀遺傳異質性和發現非編碼區域(如TE)在細胞功能中的調控作用等。
參考文獻:
Beumer N,et al. DNA hypomethylation traits define human regulatory T cells in cutaneous tissue and identify their blood recirculating counterparts. Nat Immunol. 2025 Jul 16. doi: 10.1038/s41590-025-02210-x.