
華中科技大學同濟醫學院研究團隊揭示肺動脈高壓潛在治療靶點
文章標題:Binding of Hypoxia-Induced Mitogenic Factor/RELM-β to Bone Morphogenetic Protein Receptor 2 Complex Promotes Pulmonary Hypertension
發表期刊:Arteriosclerosis, Thrombosis, and Vascular Biology (IF 7.4)
合作客戶:華中科技大學同濟醫學院基礎醫學院胡清華/肖瑞研究團隊
維真助力:
病毒產品:AAV6-HIMF、AAV6-BMP2
注射方式:氣管插管術(SD大鼠)
病毒用量:5×10^11 or 1×10^12 vg/rat
發表期刊:Arteriosclerosis, Thrombosis, and Vascular Biology (IF 7.4)
合作客戶:華中科技大學同濟醫學院基礎醫學院胡清華/肖瑞研究團隊
維真助力:
病毒產品:AAV6-HIMF、AAV6-BMP2
注射方式:氣管插管術(SD大鼠)
病毒用量:5×10^11 or 1×10^12 vg/rat
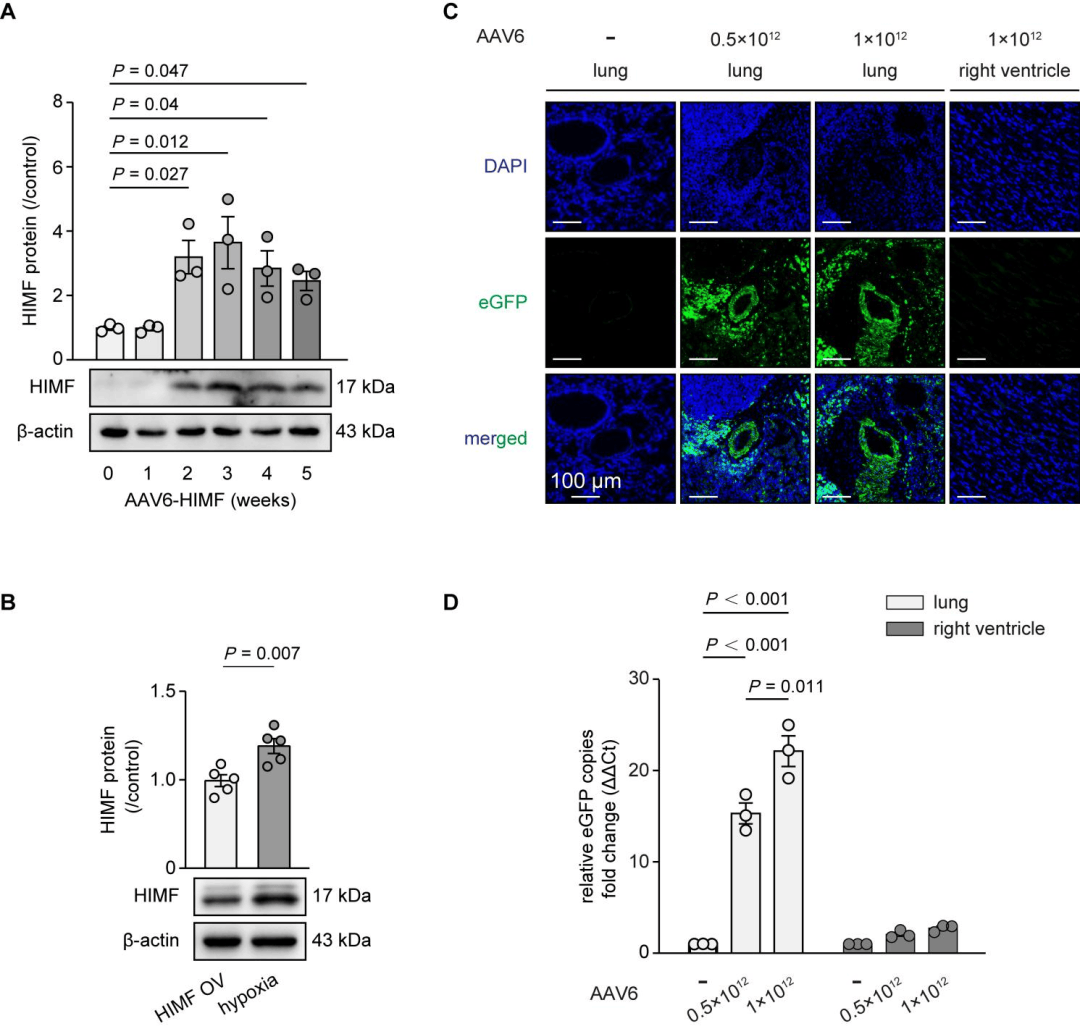
AAV6在大鼠肺組織中的遞送效率檢測
研究背景
缺氧誘導有絲分裂因子(HIMF)是一種由慢性缺氧誘導的分泌型細胞因子,可促進肺動脈高壓,但其細胞外膜受體的分子特性尚不清楚。骨形態發生蛋白受體2(BMPR2)是TGF-β/BMP家族成員,其基因突變、表達降低或功能異常,在遺傳性、特發性及慢性缺氧誘導的肺動脈高壓中均起關鍵作用,且BMPR2的激活依賴其與BMPR1家族(如BMPR1A)形成異二聚體復合物。本研究旨在篩選并鑒定HIMF的胞外受體,闡明 HIMF通過該受體調控肺動脈高壓的分子機制,探索潛在治療靶點。
缺氧誘導有絲分裂因子(HIMF)是一種由慢性缺氧誘導的分泌型細胞因子,可促進肺動脈高壓,但其細胞外膜受體的分子特性尚不清楚。骨形態發生蛋白受體2(BMPR2)是TGF-β/BMP家族成員,其基因突變、表達降低或功能異常,在遺傳性、特發性及慢性缺氧誘導的肺動脈高壓中均起關鍵作用,且BMPR2的激活依賴其與BMPR1家族(如BMPR1A)形成異二聚體復合物。本研究旨在篩選并鑒定HIMF的胞外受體,闡明 HIMF通過該受體調控肺動脈高壓的分子機制,探索潛在治療靶點。
研究結果
1.HIMF與BMPR2相互作用介導肺動脈高壓
研究團隊首先通過序列比對篩選GPR98、CaSR等候選受體,再通過HIMF抗體沉淀肺動脈膜蛋白裂解物并進行蛋白質組學分析,初篩出8個候選受體,交叉免疫沉淀和免疫印跡等證實BMPR2與HIMF存在相互作用,并且HIMF與BMPR2相關復合物的相互作用會對BMPR2活性產生負向調控,抑制其下游信號傳導,進而解除BMPR2對PASMC增殖的抑制作用,最終推動肺動脈高壓進展。隨后利用AAV6-HIMF構建大鼠HIMF過表達模型,結果顯示HIMF表達顯著升高,同時BMPR2信號通路活性降低,并伴隨肺動脈高壓典型病理表型。此外,HIMF過表達還會誘導TGF-β1表達升高和Smad2/3磷酸化增強,而BMP2過表達或FK506干預可逆轉這些效應。最后,研究人員還證實在缺氧誘導的肺動脈高壓模型中,相比于低劑量組,高劑量AAV6-BMP2可顯著減輕肺動脈高壓病理表型,并有效恢復肺動脈中BMPR2信號。安全性評估顯示,高劑量AAV6-BMP2對大鼠肝腎功能無顯著影響,且通過檢測肺和右心室中報告基因的表達,證實AAV6可特異性靶向肺組織,驗證了該基因遞送系統的特異性和安全性。
1.HIMF與BMPR2相互作用介導肺動脈高壓
研究團隊首先通過序列比對篩選GPR98、CaSR等候選受體,再通過HIMF抗體沉淀肺動脈膜蛋白裂解物并進行蛋白質組學分析,初篩出8個候選受體,交叉免疫沉淀和免疫印跡等證實BMPR2與HIMF存在相互作用,并且HIMF與BMPR2相關復合物的相互作用會對BMPR2活性產生負向調控,抑制其下游信號傳導,進而解除BMPR2對PASMC增殖的抑制作用,最終推動肺動脈高壓進展。隨后利用AAV6-HIMF構建大鼠HIMF過表達模型,結果顯示HIMF表達顯著升高,同時BMPR2信號通路活性降低,并伴隨肺動脈高壓典型病理表型。此外,HIMF過表達還會誘導TGF-β1表達升高和Smad2/3磷酸化增強,而BMP2過表達或FK506干預可逆轉這些效應。最后,研究人員還證實在缺氧誘導的肺動脈高壓模型中,相比于低劑量組,高劑量AAV6-BMP2可顯著減輕肺動脈高壓病理表型,并有效恢復肺動脈中BMPR2信號。安全性評估顯示,高劑量AAV6-BMP2對大鼠肝腎功能無顯著影響,且通過檢測肺和右心室中報告基因的表達,證實AAV6可特異性靶向肺組織,驗證了該基因遞送系統的特異性和安全性。
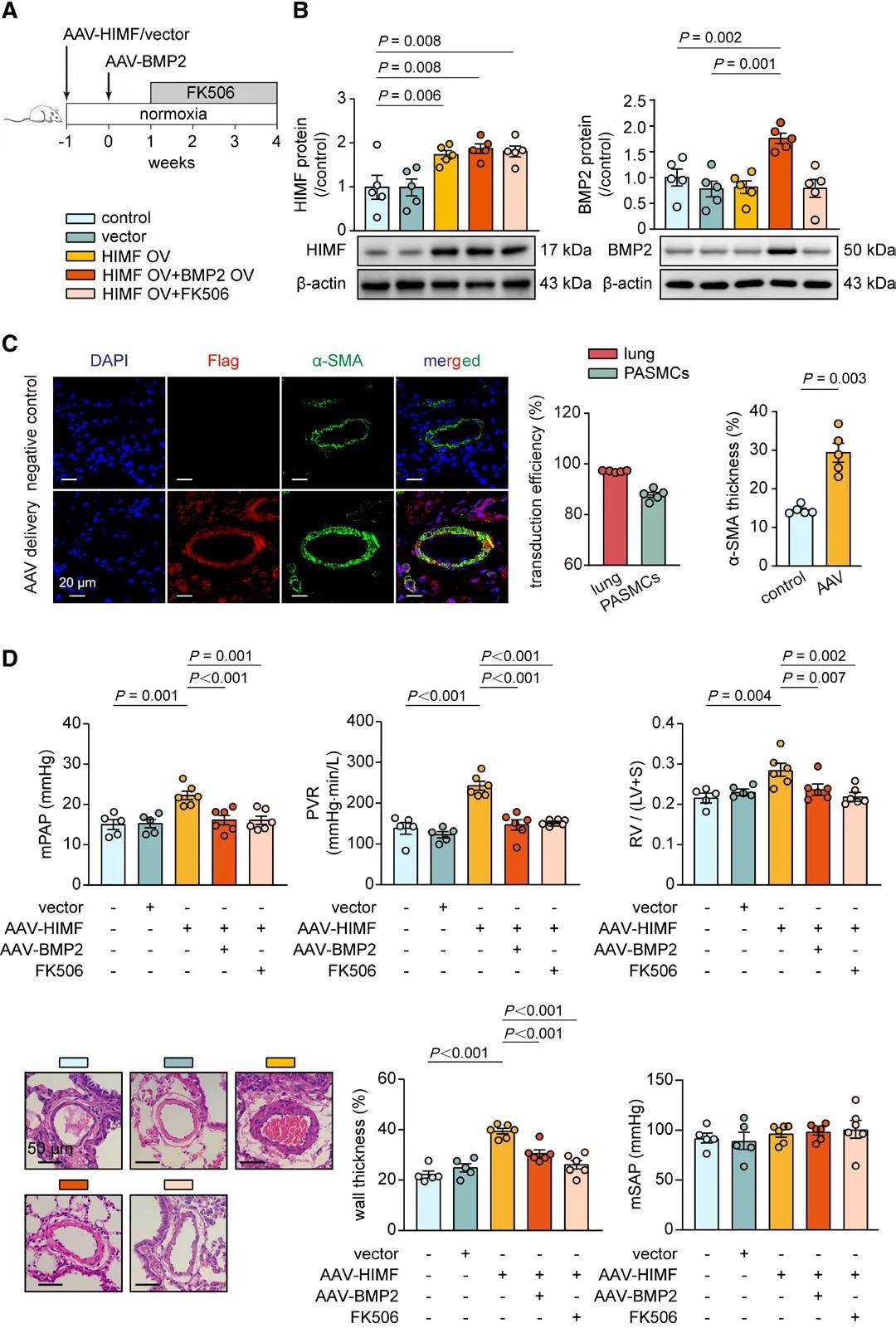
BMPR2激動劑BMP2和FK506減輕由HIMF過表達誘導的肺動脈高壓
2.阻斷HIMF-BMPR1A相互作用可減緩肺動脈高壓的發展
通過進一步的探索,研究團隊證實HIMF通過直接結合BMPR1A的DTLPF基序,競爭性破壞BMPR1A/BMPR2復合物形成,進而導致BMPR2活性降低和BMPR2表達減少。接下來,研究團隊構建了BMPR1A突變大鼠,將其胞外域54-58位的HIMF結合關鍵基序DTLPF替換為AAAAA。與野生型大鼠相比,突變大鼠在慢性缺氧條件下肺動脈高壓病理表型顯著改善,BMPR2活性得以恢復。隨后通過設計3種靶向BMPR1A DTLPF基序的阻斷肽并在原代PASMC中驗證效果,鑒定肽3在破壞HIMF與BMPR1A結合方面具有更高效率,恢復了BMPR2活性并抑制細胞增殖;此外,阻斷肽3在體內慢性缺氧及Sugen/缺氧模型中均有效緩解了肺動脈高壓。
通過進一步的探索,研究團隊證實HIMF通過直接結合BMPR1A的DTLPF基序,競爭性破壞BMPR1A/BMPR2復合物形成,進而導致BMPR2活性降低和BMPR2表達減少。接下來,研究團隊構建了BMPR1A突變大鼠,將其胞外域54-58位的HIMF結合關鍵基序DTLPF替換為AAAAA。與野生型大鼠相比,突變大鼠在慢性缺氧條件下肺動脈高壓病理表型顯著改善,BMPR2活性得以恢復。隨后通過設計3種靶向BMPR1A DTLPF基序的阻斷肽并在原代PASMC中驗證效果,鑒定肽3在破壞HIMF與BMPR1A結合方面具有更高效率,恢復了BMPR2活性并抑制細胞增殖;此外,阻斷肽3在體內慢性缺氧及Sugen/缺氧模型中均有效緩解了肺動脈高壓。
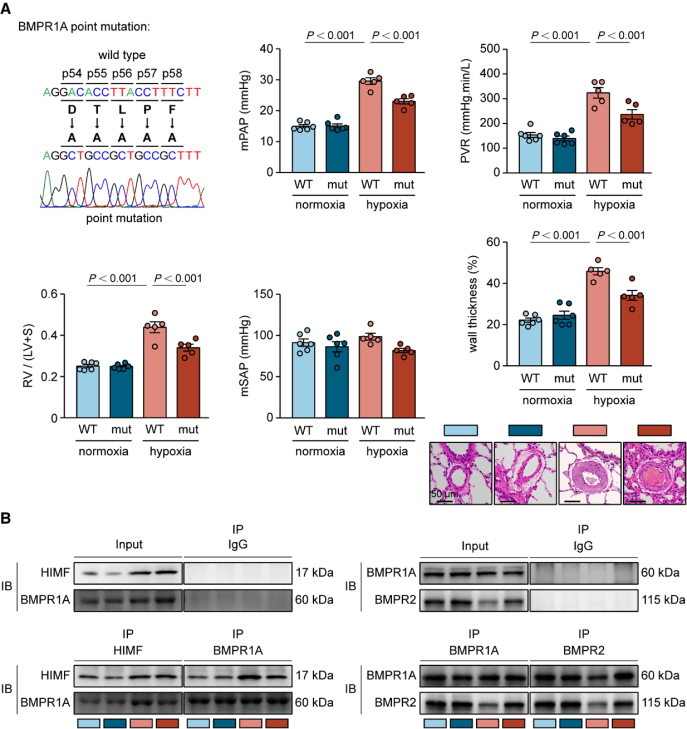
阻斷HIMF-BMPR1A相互作用可減緩肺動脈高壓的發展
研究結論
本研究表明BMPR1A是HIMF的受體,其胞外區54-58個氨基酸的結合導致BMPR1A/BMPR2異聚體復合物的破壞和BMPR2活性的降低。研究結果揭示了HIMF和BMPR1A/BMPR2信號復合物在肺動脈高壓中的病理生理作用,通過阻斷HIMF與BMPR1A的結合,提供了一種新的治療途徑來拯救BMPR2信號傳導。
本研究表明BMPR1A是HIMF的受體,其胞外區54-58個氨基酸的結合導致BMPR1A/BMPR2異聚體復合物的破壞和BMPR2活性的降低。研究結果揭示了HIMF和BMPR1A/BMPR2信號復合物在肺動脈高壓中的病理生理作用,通過阻斷HIMF與BMPR1A的結合,提供了一種新的治療途徑來拯救BMPR2信號傳導。




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com