陸軍軍醫大學第二附屬醫院團隊發現改善急性腎損傷的重要靶點
損傷相關分子模式(DAMPs)誘導的無菌性炎癥被認為是急性腎損傷(AKI)的典型特征。腎小管上皮細胞(RTECs)的質膜破裂是DAMP釋放的主要原因,神經損傷誘導蛋白1(NINJ1)被認為是質膜破裂的執行者,而其在AKI病理生理學中的作用在很大程度上尚不清楚。
2025年8月11日,陸軍軍醫大學第二附屬醫院趙景宏教授團隊在International Journal of Biological Sciences(IF 10)上發表了題為“Targeting NINJ1-Mediated Plasma Membrane Rupture in Tubular Epithelial Cell Prevents Inflammatory Response in Acute Kidney Injury”的論文。研究發現ELK1-NINJ1軸是急性腎損傷后腎小管上皮細胞質膜破裂的關鍵調節因子,表明其可能是AKI治療和預后改善的潛在靶點。
· 維真助力 - AAV·
基因信息:Ninj1:神經損傷誘導蛋白1
病毒產品及滴度:AAV9-Ksp-cadherin-shNinj1、AAV9-vector(9.64×10^13vg/ml );
實驗動物:C57BL/6J 小鼠
注射方式:腎盂注射
注射劑量:1×10^12 copies
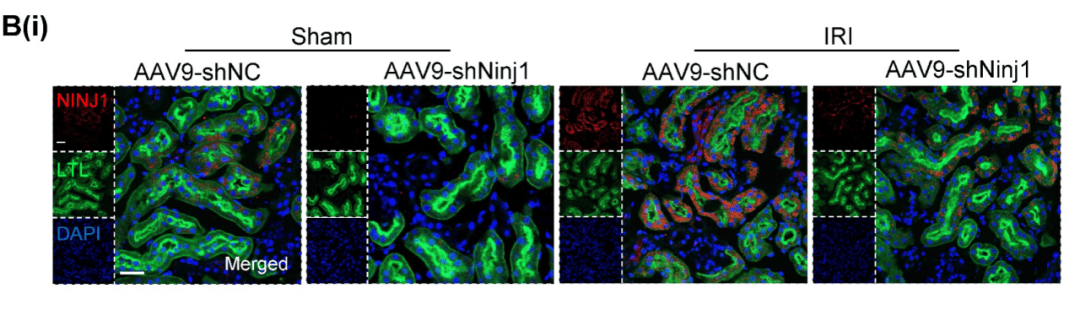 免疫熒光染色證明AAV9在RTECs中成功轉染
免疫熒光染色證明AAV9在RTECs中成功轉染
研究結果分享
1、抑制NINJ1寡聚化減弱RTECs中DAMP的釋放和炎癥反應
作者研究發現在AKI期間,RTECs中NINJ1的表達和寡聚化以及質膜破裂、DAMP釋放和炎癥反應被高度誘導。為了驗證NINJ1在RTECs中對AKI響應的具體功能及其寡聚化作用,在體外HK-2細胞中敲低NINJ1,發現NINJ1的缺失阻礙了其寡聚反應。NINJ1缺失顯著減少了由H/R誘導的氣泡解體和質膜破裂而釋放的蛋白質,表明NINJ1敲低可以減弱釋放的DAMPs。NINJ1缺失顯著降低了H/R后促炎細胞因子的釋放,相應地,敲低NINJ1也減輕了HK-2細胞的損傷。表明敲低NINJ1可以防止其寡聚化,從而進一步減輕RTECs損傷和炎癥反應。使用細胞保護劑甘氨酸處理HK-2細胞,單獨的甘氨酸處理不影響NINJ1的表達或寡聚化,但甘氨酸處理顯著抑制了H/R條件下NINJ1的寡聚化,同時伴隨沒有膜破裂,DAMPs的釋放以及HK-2細胞的炎癥減少。以上結果表明NINJ1通過誘導質膜破裂和DAMP釋放導致RTECs損傷和炎癥。
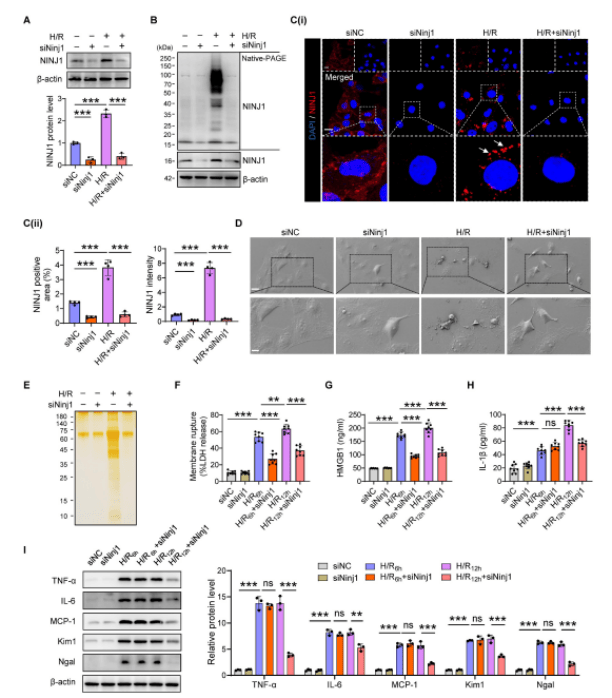 圖1. Ninj1缺失會阻斷H/R條件下DAMP的釋放和炎癥反應
圖1. Ninj1缺失會阻斷H/R條件下DAMP的釋放和炎癥反應
2、抑制NINJ1可預防AKI并改善AKI預后
為了確定NINJ1在AKI小鼠模型中的治療潛力,作者使用AAV9-shNinj1在小鼠中敲低NINJ1,觀察到AAV9-shNinj1在RTECs中成功轉染,并且使用蛋白質印跡和免疫熒光驗證了NINJ1表達的降低。與AAV9-shNC小鼠相比,AAV9-shNinj1治療顯著緩解了IRI誘導的NINJ1寡聚化,導致LDH和DAMPs釋放減少,促炎細胞因子產生減少;IRI后巨噬細胞和中性粒細胞顯著浸潤到腎間質中,而AAV9-shNinja1小鼠中這些細胞的浸潤減少。此外,敲低NINJ1降低了血清Scr和BUN水平,減輕了腎小管損傷。進一步探討了NINJ1在AKI向CKD轉變中的作用,發現AAV9-shNinj1治療顯著降低了AKI后第28天的膠原沉積、α-SMA和FN表達,從而挽救了惡化的腎功能。表明阻斷RTECs中NINJ1的表達可能是預防AKI和改善AKI預后的一種有前景的策略。
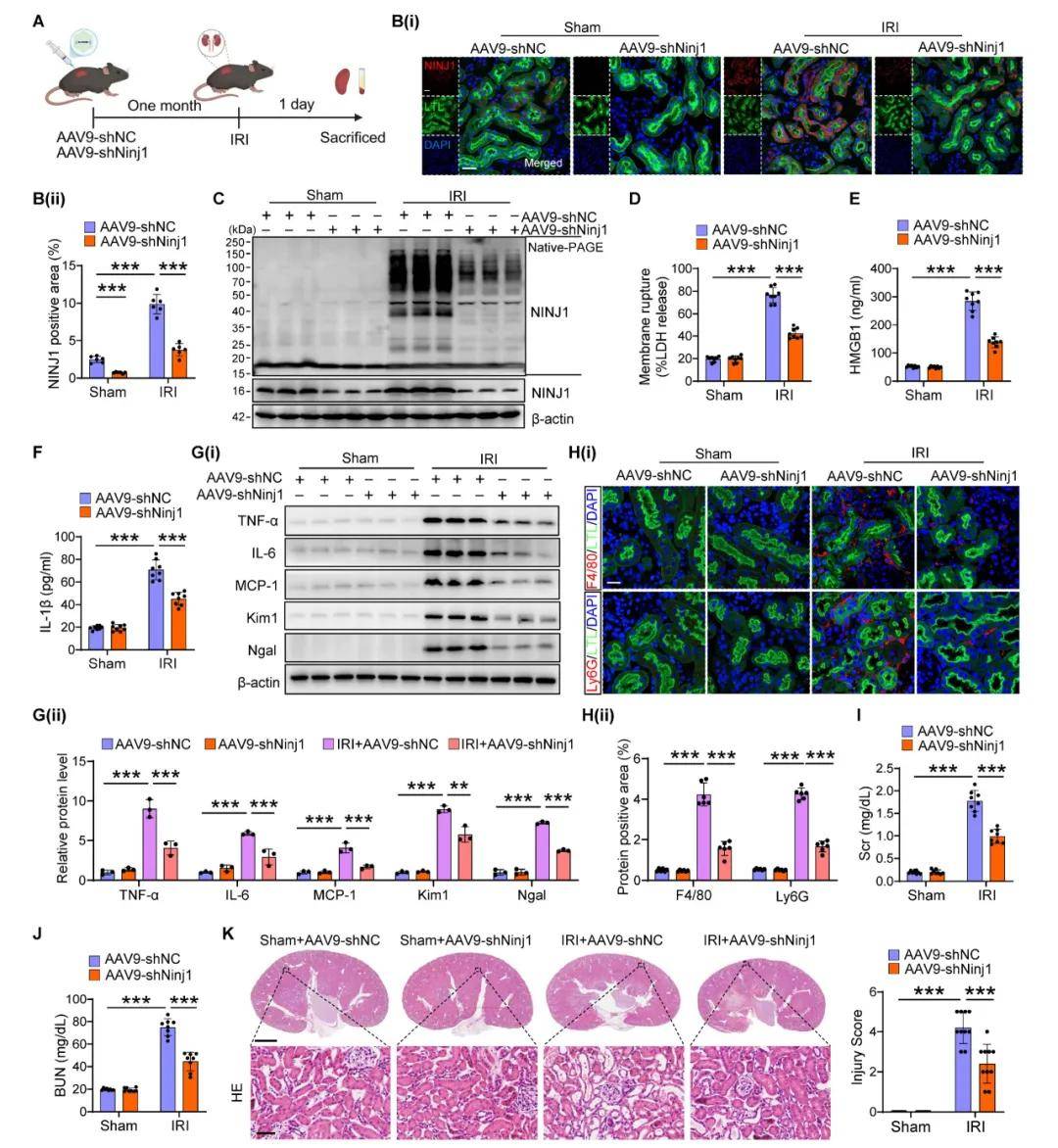 圖2. 腎小管特異性NINJ1敲除減輕IRI誘導的AKI小鼠的腎損傷
圖2. 腎小管特異性NINJ1敲除減輕IRI誘導的AKI小鼠的腎損傷
3、ELK1在Ser383磷酸化的突變減輕了NINJ1誘導的炎癥反應
作者進一步研究了NINJ1表達的分子機制,發現ELK1的Ser383磷酸化可以通過在AKI后直接結合NINJ1啟動子來調節NINJ1的轉錄表達。為了進一步確定ELK1的Ser383磷酸化位點在NINJ1誘導的DAMPs釋放和炎癥反應中的作用,作者突變了Ser383殘基并將突變體轉染到HK-2細胞中,發現Ser383的突變以及NINJ1的表達顯著消除了ELK1在Ser383的磷酸化和核轉位的顯著增加。ELK1 Ser383突變的HK-2細胞中NINJ1寡聚化受到抑制,從而保持了氣球狀形態而不是氣泡解體,并在細胞上清液中發現LDH、HMGB1、IL-1β和其他蛋白質明顯減少,減輕IRI誘導的RTEC損傷和炎癥反應。以上表明靶向ELK1的Ser383磷酸化可能限制NINJ1誘導的炎癥反應和腎損傷。作者進一步利用可以抑制ELK1在Ser383處的磷酸化的細胞穿透肽(TDE)進行體內外實驗,以防止NINJ1轉錄上調引起的炎癥,實驗表明TDE通過靶向Ser383磷酸化ELK1來緩解NINJ1介導的無菌炎癥,可能是一種有前景的治療藥物。
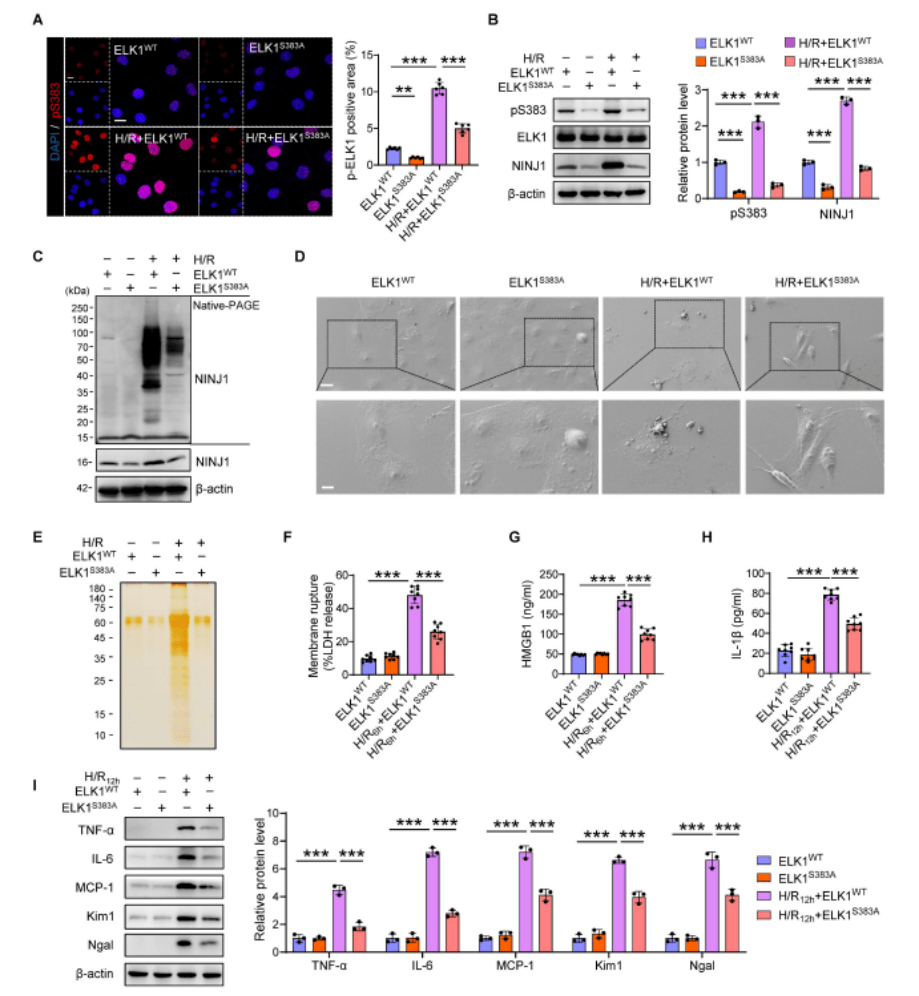 圖3. ELK1在Ser383磷酸化的突變減輕了NINJ1誘導的炎癥反應
圖3. ELK1在Ser383磷酸化的突變減輕了NINJ1誘導的炎癥反應
結論
本研究表明,RTECs中NINJ1表達的增加是AKI后質膜破裂和腎臟炎癥反應的基礎,并表明Ser383磷酸化ELK1是NINJ1表達的新轉錄調節因子。這些發現不僅為AKI的發病機制提供了深入的見解,而且表明靶向ELK1-NINJ1信號軸可能是預防這一病理過程和改善AKI預后的新策略。