利用PLM-interact擴展蛋白質語言模型以預測蛋白質-蛋白質相互作用
文章來源公眾號:AI in Graph 作者:AI in Graph

今天介紹的是發表在Nature Communications的論文: PLM-interact: extending protein language models to predict protein-protein interactions。 該論文把單蛋白語言模型擴展成“成對編碼器”,把兩條蛋白序列拼進同一上下文,聯合進行MLM + 互作二分類訓練,讓模型在注意力層面直接學“誰和誰會互作”。 結果顯示在跨物種 PPI 基準上取得SOTA,還能評估突變使互作增強/減弱,在病毒-宿主任務上也明顯優于既有方法。
1. 摘要僅根據氨基酸序列進行蛋白質結構的計算機預測已達到前所未有的精度,但預測蛋白質-蛋白質相互作用仍然是一個挑戰。本文,作者評估了常用于蛋白質折疊的蛋白質語言模型 (PLM) 重新訓練用于蛋白質-蛋白質相互作用預測的能力。現有的利用 PLM 的模型使用預訓練的 PLM 特征集,忽略了蛋白質之間的物理相互作用。作者提出了 PLM-interact,它超越了單個蛋白質,通過聯合編碼蛋白質對來學習它們之間的關系,類似于自然語言處理中的下一句預測任務。該方法在廣泛采用的跨物種蛋白質-蛋白質相互作用預測基準中取得了最佳性能:基于人類數據進行訓練,并在小鼠、蒼蠅、線蟲、大腸桿菌和酵母上進行測試。此外,作者開發了一種 PLM-interact 的微調方法,以檢測突變對相互作用的影響。最后,作者報告該模型在蛋白質水平上預測病毒-宿主相互作用方面優于現有方法。作者的工作表明,大型語言模型可以擴展,僅從生物分子序列中就可以了解生物分子之間的復雜關系。
2. 引言
僅憑序列預測蛋白結構已十分成熟,但要“只看序列”判定兩條蛋白是否互作(PPI)仍很難:實驗標注稀缺昂貴、跨物種分布差異顯著,若數據拆分不嚴還會因相似性“泄漏”而高估性能。更關鍵的是,主流序列法多沿用“雙塔/兩段式”范式:分別編碼兩條序列,末端再用小分類頭“猜”是否互作;這種流程讓語言模型始終以“單蛋白”為基本單位,并不“意識到”兩條鏈彼此成對,真正的跨鏈線索被推遲到末端分類器處理,難以在跨物種與低樣本場景中穩健泛化。
PLM-interact 的出發點是把“配對關系”直接放進語言模型的上下文:將兩條蛋白一次性輸入同一個 Transformer,以跨編碼(cross-encoder)結構在編碼階段就讓注意力對齊跨蛋白殘基;訓練上聯合遮蓋語言模型(MLM)與互作二分類,通過權重與遮蓋比例的系統搜索,在保留語言理解能力的同時,迫使模型學習“哪對殘基彼此有關”,從而把“互作判斷”前移到表示學習之中,減輕末端分類頭的容量約束。
在嚴格的人類訓練→多物種測試(鼠、蠅、蟲、酵母、大腸桿菌)的基準上,PLM-interact 在 AUPR 上普遍領先,并表現出更穩定的正樣本區分能力;在去重控相似度的人類無泄漏數據、以及突變效應與病毒-宿主互作任務中,同樣表現穩健且可通過端到端微調進一步提升區分度。相較傳統“雙塔”,這種“把配對放進上下文、讓注意力跨鏈工作”的范式,為僅憑序列的 PPI 預測提供了更自然的建模路徑,并為后續融合結構、網絡與功能注釋等多模態信息留下了清晰接口。
3. 方法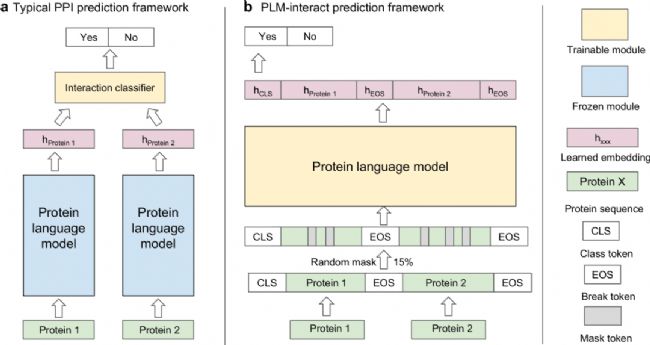
傳統“雙塔”框架 vs. PLM-interact 跨編碼框架示意
3.1 框架與輸入
作者以 ESM-2(默認 650M)為基座,將兩條蛋白序列在同一個 Transformer中跨鏈路同時編碼(cross-encoder)。標準輸入序列為
ESM-2 編碼得到各 token 的輸出嵌入,用 [CLS] 向量經一層前饋網絡后接 Sigmoid 得到互作概率:
3.2 訓練目標與技巧模型以遮蓋語言模型(MLM)與二分類聯合優化,單樣本損失為
作者系統比較權重后,采用 分類:MLM = 1:10 的比例,并配合 15% 隨機遮蓋;該設置在多物種基準上綜合最優。
為容納兩條序列,放寬總長度閾值(例如 STRING V12 訓練對的總長閾值 2101),并對每個訓練對雙向喂入 與 以增強順序不變性。
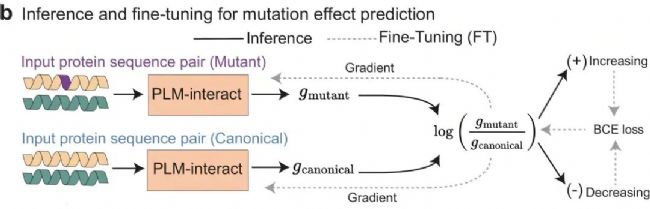
3.3 突變效應預測給定某 PPI,改變其中一個蛋白為突變體。首先用式(2)分別得到野生型與突變型的互作概率 。定義對數概率比
以其符號預測增強(+)或削弱(−)的二分類標簽,并將 (lr) 作為輸入信號、用交叉熵損失端到端反向傳播到所有層進行微調(只微調分類頭明顯不如全模型微調)。該流程使用 IntAct 的增強/削弱互作注釋(共 6,979 條),顯著提升 AUPR/AUROC。
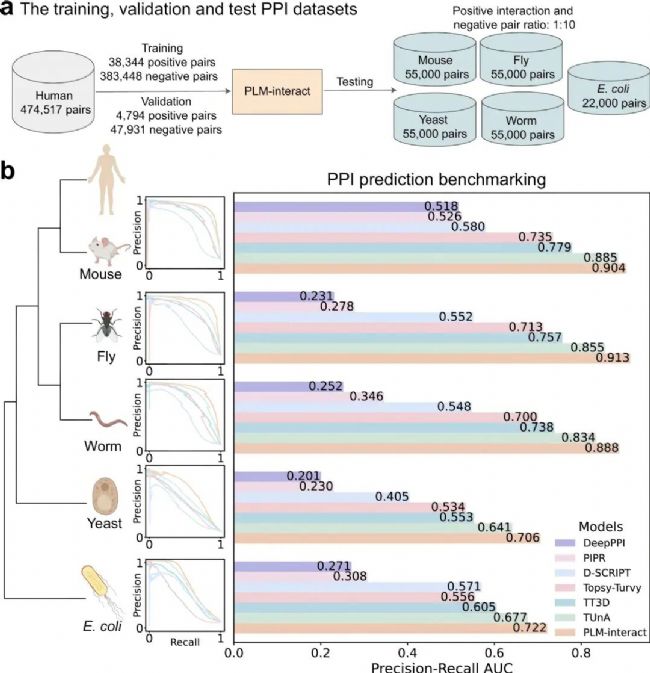
4. 實驗
4.1 跨物種基準
基準采用 1:10(正:負) 的配比;人類訓練集含 38,344 條正樣本(驗證集 4,794 正),五個測試物種各含 5,000 條正樣本(E. coli 為 2,000 正)。在該設置下,PLM-interact 的 AUPR 在全部物種領先:例如酵母 AUPR=0.706(較 TUnA 的 0.641 提升 10%)、E. coli AUPR=0.722(較 TUnA 提升 7%);作者指出優勢主要來自對正樣本賦予更高互作概率,而且交換鏈順序后 AUPR 基本不變,顯示推斷對順序魯棒。
4.2 無泄漏人類數據集
在 Bernett 去重控相似度的人類基準上,PLM-interact 與 TUnA 的 AUPR≈0.69 / AUROC≈0.70 基本持平;但當采用中性閾值(0.5)做最終分類時,PLM-interact 的 F1 與 Recall 更高(召回+9%,精度與 TUnA 相當),表明其對真陽性更敏感。
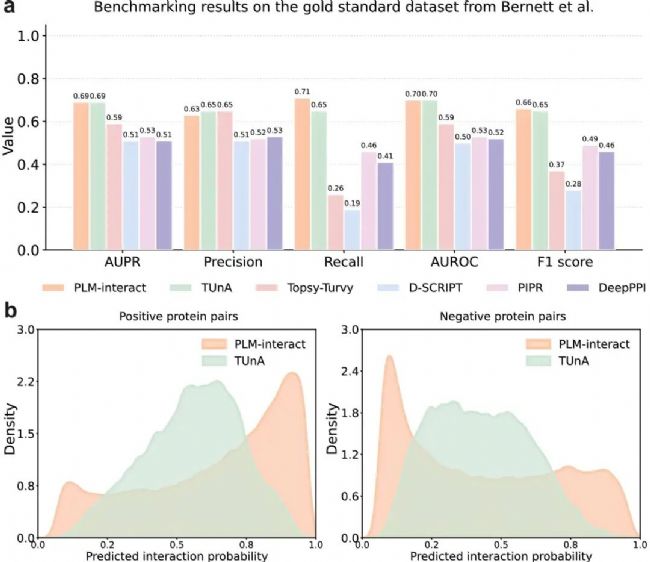
4.3 突變效應(IntAct 注釋的增強/削弱互作)
使用 IntAct 的突變注釋(共 6,979 條,增互作/減互作二類)評估“零樣本”與“微調”。零樣本下所有方法接近隨機;對 PLM-interact 端到端微調全部層后,AUPR 提升約 150%、AUROC 提升 36%,并給出兩個成功案例:MCM7-Y600E(增強)與 ISCU-N151A(減弱),模型均正確判定方向。
4.4 病毒–宿主 PPI(HPIDB 派生)
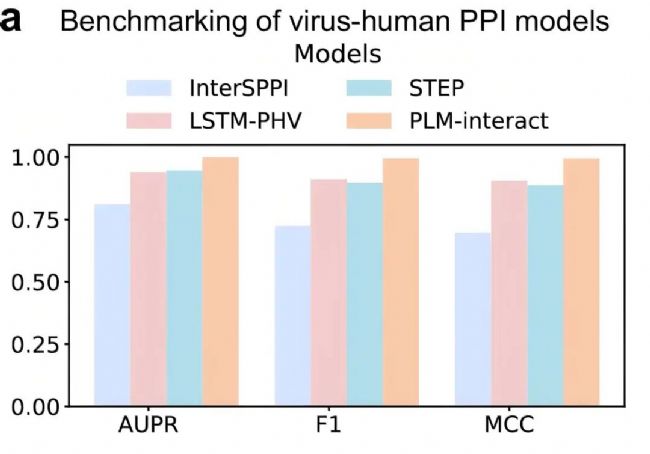
在 Tsukiyama 等構建的 HPIDB 3.0 派生數據(共 22,383 條 PPI,1:10 配比)上,對比 STEP(ProtBERT 特征)、LSTM-PHV、InterSPPI:PLM-interact 在 AUPR、F1、MCC 上全面領先,相對 STEP 的提升分別為 +5.7% AUPR、+10.9% F1、+11.9% MCC;作者同時展示了若干已有實驗結構的病毒-人互作示例。
4.5 消融與訓練技巧
在 0%、7%、15%、22%、30% 中,15% 是唯一在統計上顯著優于“僅二分類(0%)”的設定。損失權重: 在 ESM-2-650M 下,分類:MLM=1:10 綜合最佳(E. coli AUPR 相對第二名+4.3%)。順序不變性: 測試時交換鏈順序,AUPR 與分布幾乎不變。
5. 結論與未來展望
PLM-interact 的關鍵在于把 PPI 從“兩段獨白”變為“同域對話”:將兩條序列拼接進同一上下文,跨蛋白注意力直接對齊殘基依賴,配合“二分類+MLM(1:10)”的聯合目標與 ESM-2(650M) 初始化,在跨物種、無泄漏、突變效應與病毒-宿主任務上穩健領先。實際應用上,它可用于新物種互作網絡冷啟動、突變增/減效的快速判別,以及病原-宿主互作識別與藥靶發現。面向未來,值得繼續沿著多模態融合(序列+結構/網絡)、更長上下文與多實體協同建模、以及輕量化與不確定性校準等方向推進,以在保持推理效率的同時提升可解釋性與部署可用性。