
蛋白質組學基礎:定量方法及原理
定量蛋白質組學旨在對生物體、組織、細胞等樣本中的蛋白質進行定量分析,研究其在不同生理病理狀態、不同發育階段或不同環境條件下的表達水平變化,以深入了解蛋白質功能、揭示生命活動規律和疾病發生發展機制。基于質譜的蛋白質組學定量目前已成為主流方法,在標志物發現和驗證研究中發揮重要作用。本期推送我們就來為大家介紹一下常用的幾種定量方法。
質譜蛋白質組定量方法可分為非標記定量、標記定量和靶向定量三個大類。非標記定量包括 DDA和DIA,前者依據一級質譜峰面積定量,但低豐度蛋白易漏檢;后者對選定質荷比范圍內的所有離子同時碎裂和掃描,并且基于MS2完成定性和定量,更準確、重現性更好。標記定量包括基于 MS2/MS3 報告離子定量的 TMT和iTRAQ,可同時標記多個樣本,還有基于MS1代謝標記定量的 SILAC,僅適用于體外培養細胞樣本。前述定量方法均用來在復雜樣本的全蛋白組中篩選潛在差異表達蛋白,而靶向定量的MRM和PRM 則針對特定蛋白質或肽段進行掃描和定量分析,特異性強、準確性高,適用于后期標志物驗證。
01.非標記定量
DDA和DIA都屬于非標記定量(LFQ),無需對蛋白質進行額外標記,主要依據質譜檢測到的信號強度或峰面積來推斷蛋白質含量。該方法實驗操作相對簡便,能避免標記過程引入的偏差,不受標記數量的限制,適用于大規模蛋白質組學研究,但容易受到質譜分析過程中的各種因素影響,需做好嚴格的質控。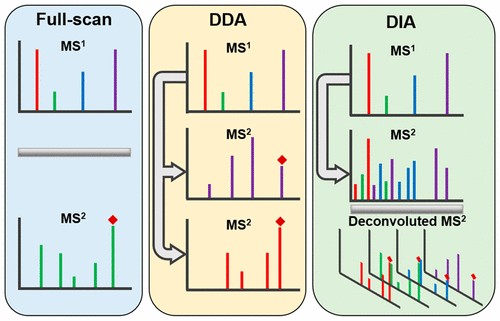
圖1 DDA和DIA采集原理示意[1]
1.1 DDA(數據依賴型采集)
DDA是最常用的非標記定量方法之一,主要通過比較不同樣本中相同蛋白質的一級質譜峰強度或峰面積來實現。PEAKS軟件的DDA LFQ定量是基于肽段特征峰(Peptide feature)面積的,feature指的是同一肽段的所有同位素峰(圖2)。通過對比不同樣本間定量值的高低判定其相對含量(圖3)。蛋白定量值默認使用打分top 3的unique肽段定量值加和,在某些樣本中的定量值高則表明在該樣本中的表達量相對較高。
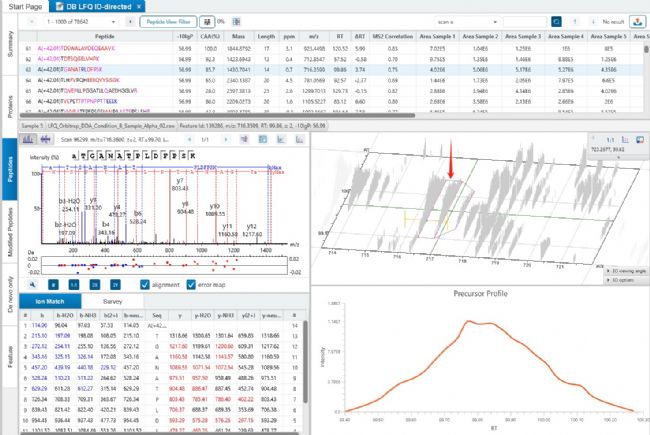 圖2 peptide feature示例
圖2 peptide feature示例
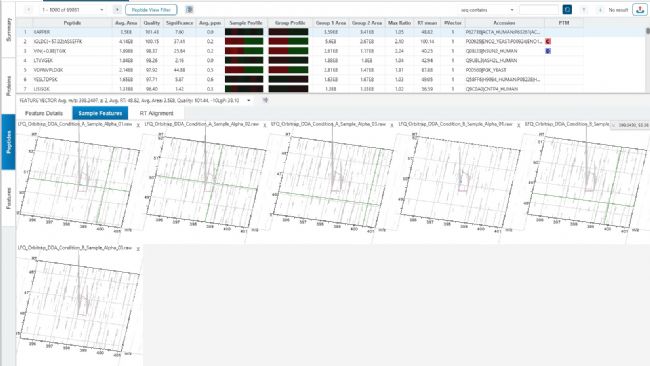 圖3 同一肽段在不同樣本間的特征峰分布
圖3 同一肽段在不同樣本間的特征峰分布
DDA的實驗和數據分析相對比較容易,并且適合未知的翻譯后修飾和潛在突變位點分析,成本低,不過也存在一定局限性。由于其是基于信號強度選擇母離子進行二級碎裂,低豐度肽段的母離子可能因信號響應過低而無法被選中,導致定量缺失,影響蛋白質定量的準確性和重現性。
1.2 DIA(數據非依賴型采集)
DIA技術是為克服DDA的局限性而發展起來的。在DIA掃描模式下,儀器不再選擇性地對特定離子進行碎裂,而是對選定質荷比范圍內的所有離子同時進行碎裂和掃描。常規DIA方法是將整個質譜掃描范圍劃分為多個連續的窗口,依次對每個窗口內的離子進行碎裂和MS2掃描,獲得所有離子的二級譜。DIA的優勢在于能夠無遺漏、無偏差地檢測樣本中的所有離子,定性和定量均基于二級譜完成,蛋白定量不是簡單的肽段定量值的加和,而是采用最小二乘法,將所有肽段納入計算(圖4)。
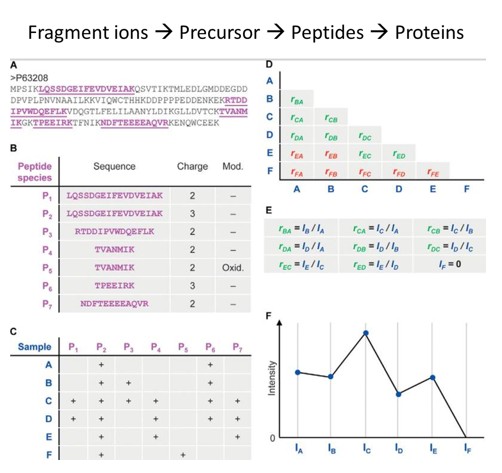
圖4 DIA定量邏輯[2]
DIA比DDA定量結果更準確、重復性更好,尤其適用于復雜樣本的深度蛋白質組定性和定量分析。但DIA數據的譜圖極為復雜,因此對分析軟件和算法的要求也就更高。PEAKS Studio和PEAKS Online支持DIA數據的定性和定量分析,并且全面兼容譜圖庫搜索(spectral library)、直接數據庫搜索(DIA database search)和DIA de novo,根據分析需求直接選擇相應的分析工作流即可(圖5)。
 圖5 PEAKS中的DIA數據分析工作流
圖5 PEAKS中的DIA數據分析工作流
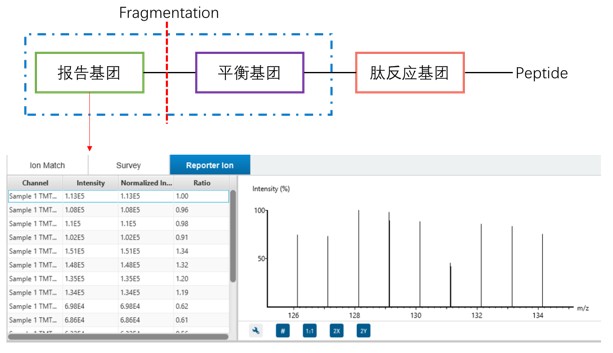
圖6 報告離子定量示意
報告離子定量技術的優點是能夠同時對多個樣本(TMT最多可同時標記18個樣本,iTRAQ通常可標記4-8個樣本)進行定量分析,通量高,靈敏度也較好。然而,標記過程較為繁瑣,成本相對較高。
2.2 基于MS1的代謝標記定量
穩定同位素標記氨基酸在細胞培養中的應用(SILAC)是典型的基于MS1的代謝標記定量方法。在細胞培養過程中,向培養基中添加含有穩定同位素(如13C、15N等)標記的必需氨基酸(如精氨酸、賴氨酸)。細胞在生長代謝過程中,會將這些標記的氨基酸整合到新合成的蛋白質中。經過多代培養后,細胞內新合成的蛋白質幾乎都被穩定同位素標記。將標記細胞與未標記細胞(對照組)的蛋白質提取物混合,進行質譜分析。在MS1掃描時,由于標記和未標記蛋白質含有不同質量的氨基酸,它們的質荷比會出現差異,形成具有特定質量差的峰對。通過比較這些峰對的強度,就可以準確計算出標記樣本和未標記樣本中蛋白質的相對含量(圖7)。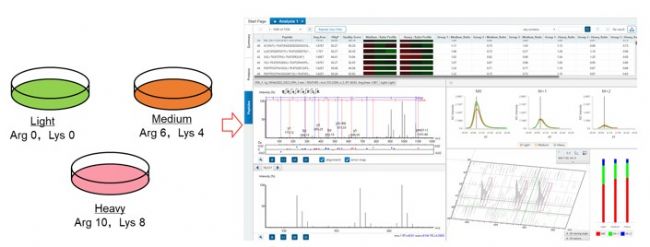
圖7 SILAC定量示意
SILAC的優勢在于標記過程在細胞培養階段完成,標記均勻且對蛋白質的后續處理影響較小,定量準確性高。但該方法標記通道有限,僅適用于能夠在體外培養的細胞樣本,對于組織樣本或難以培養的細胞則無法使用,應用范圍存在一定限制,標記周期長,試劑成本也有所增加。
03.靶向定量
質譜蛋白質組定量方法可分為非標記定量、標記定量和靶向定量三個大類。非標記定量包括 DDA和DIA,前者依據一級質譜峰面積定量,但低豐度蛋白易漏檢;后者對選定質荷比范圍內的所有離子同時碎裂和掃描,并且基于MS2完成定性和定量,更準確、重現性更好。標記定量包括基于 MS2/MS3 報告離子定量的 TMT和iTRAQ,可同時標記多個樣本,還有基于MS1代謝標記定量的 SILAC,僅適用于體外培養細胞樣本。前述定量方法均用來在復雜樣本的全蛋白組中篩選潛在差異表達蛋白,而靶向定量的MRM和PRM 則針對特定蛋白質或肽段進行掃描和定量分析,特異性強、準確性高,適用于后期標志物驗證。
01.非標記定量
DDA和DIA都屬于非標記定量(LFQ),無需對蛋白質進行額外標記,主要依據質譜檢測到的信號強度或峰面積來推斷蛋白質含量。該方法實驗操作相對簡便,能避免標記過程引入的偏差,不受標記數量的限制,適用于大規模蛋白質組學研究,但容易受到質譜分析過程中的各種因素影響,需做好嚴格的質控。
圖1 DDA和DIA采集原理示意[1]
1.1 DDA(數據依賴型采集)
DDA是最常用的非標記定量方法之一,主要通過比較不同樣本中相同蛋白質的一級質譜峰強度或峰面積來實現。PEAKS軟件的DDA LFQ定量是基于肽段特征峰(Peptide feature)面積的,feature指的是同一肽段的所有同位素峰(圖2)。通過對比不同樣本間定量值的高低判定其相對含量(圖3)。蛋白定量值默認使用打分top 3的unique肽段定量值加和,在某些樣本中的定量值高則表明在該樣本中的表達量相對較高。
圖2 peptide feature示例圖3 同一肽段在不同樣本間的特征峰分布DDA的實驗和數據分析相對比較容易,并且適合未知的翻譯后修飾和潛在突變位點分析,成本低,不過也存在一定局限性。由于其是基于信號強度選擇母離子進行二級碎裂,低豐度肽段的母離子可能因信號響應過低而無法被選中,導致定量缺失,影響蛋白質定量的準確性和重現性。
1.2 DIA(數據非依賴型采集)
DIA技術是為克服DDA的局限性而發展起來的。在DIA掃描模式下,儀器不再選擇性地對特定離子進行碎裂,而是對選定質荷比范圍內的所有離子同時進行碎裂和掃描。常規DIA方法是將整個質譜掃描范圍劃分為多個連續的窗口,依次對每個窗口內的離子進行碎裂和MS2掃描,獲得所有離子的二級譜。DIA的優勢在于能夠無遺漏、無偏差地檢測樣本中的所有離子,定性和定量均基于二級譜完成,蛋白定量不是簡單的肽段定量值的加和,而是采用最小二乘法,將所有肽段納入計算(圖4)。
圖4 DIA定量邏輯[2]
DIA比DDA定量結果更準確、重復性更好,尤其適用于復雜樣本的深度蛋白質組定性和定量分析。但DIA數據的譜圖極為復雜,因此對分析軟件和算法的要求也就更高。PEAKS Studio和PEAKS Online支持DIA數據的定性和定量分析,并且全面兼容譜圖庫搜索(spectral library)、直接數據庫搜索(DIA database search)和DIA de novo,根據分析需求直接選擇相應的分析工作流即可(圖5)。
圖5 PEAKS中的DIA數據分析工作流
02.標記定量
標記定量是通過對蛋白質或肽段進行化學標記,使不同樣本中的蛋白質帶有可區分的標記物,從而在質譜檢測時更精準地進行定量分析。該方法能有效提高定量的準確性和靈敏度,減少樣本間的誤差。
2.1 基于MS2/MS3的報告離子定量
常用的基于MS2/MS3報告離子定量技術有TMT和iTRAQ。TMT和iTRAQ試劑均由報告基團、平衡基團和反應基團組成。反應基團可與蛋白質或肽段的特定基團(如賴氨酸殘基的氨基)發生共價結合,實現標記。標記后的不同樣本混合進行質譜分析。在MS1掃描時,由于標記后的肽段帶有相同質量的標簽,所以具有相同的質荷比,無法區分不同樣本來源的肽段。但在MS2掃描時,肽段發生碎裂,報告基團從標簽上脫落,產生具有特征質量的報告離子。不同樣本的報告離子質量不同,通過檢測這些報告離子的強度,就能計算出不同樣本中相應蛋白質的相對含量(圖6)。部分情況下,還可以進行TMT MS3掃描,進一步提高定量的準確性,減少干擾。
標記定量是通過對蛋白質或肽段進行化學標記,使不同樣本中的蛋白質帶有可區分的標記物,從而在質譜檢測時更精準地進行定量分析。該方法能有效提高定量的準確性和靈敏度,減少樣本間的誤差。
2.1 基于MS2/MS3的報告離子定量
常用的基于MS2/MS3報告離子定量技術有TMT和iTRAQ。TMT和iTRAQ試劑均由報告基團、平衡基團和反應基團組成。反應基團可與蛋白質或肽段的特定基團(如賴氨酸殘基的氨基)發生共價結合,實現標記。標記后的不同樣本混合進行質譜分析。在MS1掃描時,由于標記后的肽段帶有相同質量的標簽,所以具有相同的質荷比,無法區分不同樣本來源的肽段。但在MS2掃描時,肽段發生碎裂,報告基團從標簽上脫落,產生具有特征質量的報告離子。不同樣本的報告離子質量不同,通過檢測這些報告離子的強度,就能計算出不同樣本中相應蛋白質的相對含量(圖6)。部分情況下,還可以進行TMT MS3掃描,進一步提高定量的準確性,減少干擾。
圖6 報告離子定量示意
報告離子定量技術的優點是能夠同時對多個樣本(TMT最多可同時標記18個樣本,iTRAQ通常可標記4-8個樣本)進行定量分析,通量高,靈敏度也較好。然而,標記過程較為繁瑣,成本相對較高。
2.2 基于MS1的代謝標記定量
穩定同位素標記氨基酸在細胞培養中的應用(SILAC)是典型的基于MS1的代謝標記定量方法。在細胞培養過程中,向培養基中添加含有穩定同位素(如13C、15N等)標記的必需氨基酸(如精氨酸、賴氨酸)。細胞在生長代謝過程中,會將這些標記的氨基酸整合到新合成的蛋白質中。經過多代培養后,細胞內新合成的蛋白質幾乎都被穩定同位素標記。將標記細胞與未標記細胞(對照組)的蛋白質提取物混合,進行質譜分析。在MS1掃描時,由于標記和未標記蛋白質含有不同質量的氨基酸,它們的質荷比會出現差異,形成具有特定質量差的峰對。通過比較這些峰對的強度,就可以準確計算出標記樣本和未標記樣本中蛋白質的相對含量(圖7)。
圖7 SILAC定量示意
SILAC的優勢在于標記過程在細胞培養階段完成,標記均勻且對蛋白質的后續處理影響較小,定量準確性高。但該方法標記通道有限,僅適用于能夠在體外培養的細胞樣本,對于組織樣本或難以培養的細胞則無法使用,應用范圍存在一定限制,標記周期長,試劑成本也有所增加。
03.靶向定量
靶向定量是對預先設定的特定母離子進行定量分析,具有高靈敏度、高特異性和準確性的特點,是蛋白質組學研究中發現的潛在生物標志物驗證的重要方法。如圖8A所示,多重反應監測(MRM)及平行反應監測(PRM)是常用的靶向定量方法。在MRM模式下,儀器首先選擇目標肽段的母離子(特定質荷比的離子)進行檢測(Q1選擇),母離子在碰撞室中碎裂后,儀器再選擇特定的碎片離子(Q3選擇)進行監測。通過監測母離子到特定碎片離子的反應,能夠特異性地檢測目標肽段,排除其他干擾。PRM技術則是在MRM基礎上發展而來,它利用高分辨率質譜,能夠同時監測多個目標肽段的所有碎片離子,獲得更全面的信息,提高定量的準確性和可靠性(圖8B)。靶向定量需要預先了解目標肽段母離子的信息,如氨基酸序列、m/z等,通過針對性地優化檢測條件,實現對目標肽段和蛋白的精確定量。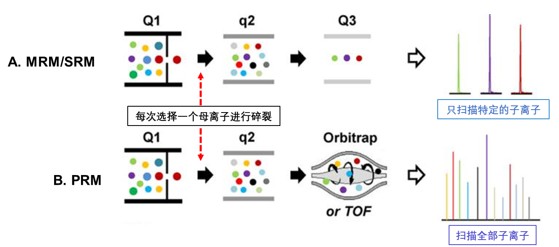
圖8 靶向定量示意
04.參考文獻
[1]. Guo J, Huan T. Comparison of Full-Scan, Data-Dependent, and Data-Independent Acquisition Modes in Liquid Chromatography-Mass Spectrometry Based Untargeted Metabolomics. Anal Chem. 2020 Jun 16;92(12):8072-8080. doi: 10.1021/acs.analchem.9b05135. Epub 2020 May 27. PMID: 32401506.
[2].Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014 Sep;13(9):2513-26. doi: 10.1074/mcp.M113.031591. Epub 2014 Jun 17. PMID: 24942700; PMCID: PMC4159666.
作為生物信息學的領軍企業,BSI專注于蛋白質組學和生物藥領域,通過機器學習和先進算法提供世界領先的質譜數據分析軟件和蛋白質組學服務解決方案,以推進生物學研究和藥物發現。我們通過基于AI的計算方案,為您提供對蛋白質組學、基因組學和醫學的卓越洞見。旗下著名的PEAKS®️系列軟件在全世界擁有數千家學術和工業用戶,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽組發現服務和抗體綜合表征服務等。聯系方式:021-60919891;sales-china@bioinfor.com
[1]. Guo J, Huan T. Comparison of Full-Scan, Data-Dependent, and Data-Independent Acquisition Modes in Liquid Chromatography-Mass Spectrometry Based Untargeted Metabolomics. Anal Chem. 2020 Jun 16;92(12):8072-8080. doi: 10.1021/acs.analchem.9b05135. Epub 2020 May 27. PMID: 32401506.
[2].Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014 Sep;13(9):2513-26. doi: 10.1074/mcp.M113.031591. Epub 2014 Jun 17. PMID: 24942700; PMCID: PMC4159666.
作為生物信息學的領軍企業,BSI專注于蛋白質組學和生物藥領域,通過機器學習和先進算法提供世界領先的質譜數據分析軟件和蛋白質組學服務解決方案,以推進生物學研究和藥物發現。我們通過基于AI的計算方案,為您提供對蛋白質組學、基因組學和醫學的卓越洞見。旗下著名的PEAKS®️系列軟件在全世界擁有數千家學術和工業用戶,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽組發現服務和抗體綜合表征服務等。聯系方式:021-60919891;sales-china@bioinfor.com




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com