
PEAKS 培訓課:AI模型在高通量蛋白質組學中的應用

BSI 專注于蛋白質組學與生物藥領域,依托機器學習與先進算法開發世界領先的質譜數據分析軟件,助力加速生物學研究與藥物研發進程。
我們有幸受邀于10月10-11日承辦PEAKS Training Workshop,為您帶來深度、前沿的專業培訓,帶您走進深度蛋白質組學的世界!歡迎各位老師的參與!
會議注冊地址:http://aohupo2025.com/register
(注冊時請選擇 PEAKS Online Training)
BSI, dedicated to proteomics and biopharmaceuticals, provides world-leading mass spectrometry data analysis software powered by machine learning and advanced algorithms to accelerate biological research and drug discovery.
We are honored to be invited to host the PEAKS Training Workshop on October 10–11, delivering in-depth, cutting-edge training to take you into the world of Deep Proteomics! We warmly welcome your participation!
Registered address:http://aohupo2025.com/register
(Remember to Choose PEAKS OnlineTraining)
本次為期兩天的 PEAKS 培訓課程,將介紹 AI 模型在高通量蛋白質組學中的應用;展示如何針對多種互補的質譜方法,結合數據庫搜索和從頭測序的數據分析,達到在proteoform水平上的深度覆蓋。無論是對于剛剛進入AI賦能的蛋白質組領域的新手,還是有經驗的大隊列樣本的蛋白質組學研究,都非常適合參加這個培訓。
In PEAKS Online training workshop, we are going to introduce AI models for high throughput proteomics with deep coverage at proteoform level, by integrate alternative MS approaches with complementary database search and de novo sequencing data analysis. This training is highly suitable for both newcomers who have just entered the AI empowered proteomics field and experienced researchers engaged in proteomics studies of large cohort samples.
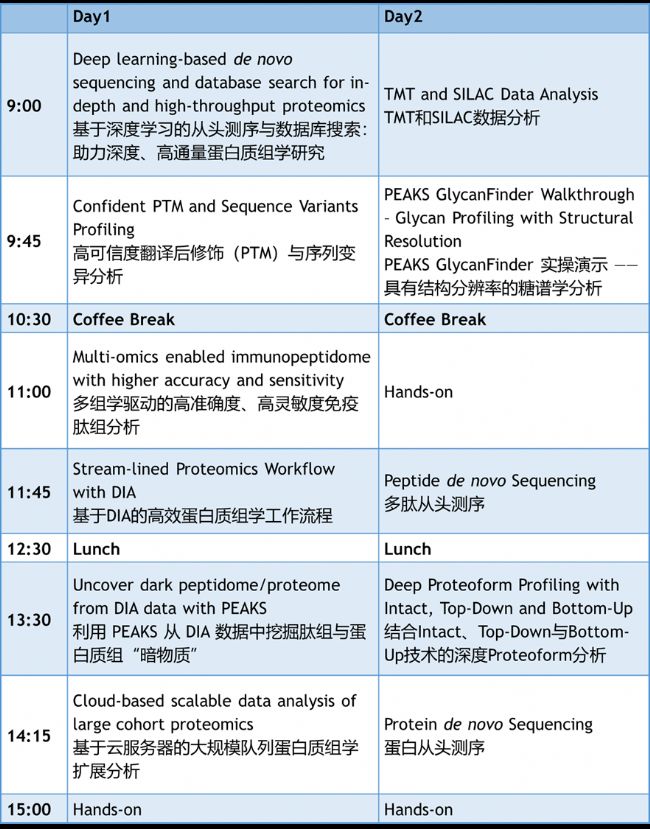
# 基于深度學習的從頭測序與數據庫搜索:助力深度、高通量蛋白質組學研究 Deep learning-based de novo sequencing and database search for in-depth and high-throughput proteomics
整合數據庫搜索與從頭測序技術,并結合多種酶解策略,可實現蛋白質組的深度覆蓋。覆蓋度可達到1.5萬個蛋白組中的100萬條獨特肽段,平均序列覆蓋率約為80%。
Integrating database search and de novo sequencing and combining multiple enzyme digests facilitated deeper proteome coverage up to a million unique peptides from 15,000 protein groups, with a median sequence coverage of approximately 80%.
# 高可信度翻譯后修飾(PTM)與序列變異分析 Confident PTM and Sequence Variants Profiling借助基于人工智能驅動的評分模型,PEAKS在鑒定翻譯后修飾(PTM)與序列變異時可實現超高可信度,有效解決了在低豐度修飾或序列變異檢測中常見的靈敏度與準確性問題。
Leveraging AI-powered scoring models, PEAKS achieves ultra-high confidence in identifying post-translational modifications (PTMs) and sequence variants, addressing the common issue of sensitivity and accuracy in low-abundance modification or variant detection.
#多組學驅動的高準確度、高靈敏度免疫肽組分析 Multi-omics enabled immunopeptidome with higher accuracy and sensitivity將LC-MS/MS數據與下一代測序(NGS)結果相結合,PEAKS 可實現全面的免疫肽組分析, 突破單一技術手段的局限,確保精準鑒定主要組織相容性復合體(MHC)結合肽,為癌癥免疫治療研究提供支持。
Integrating LC-MS/MS data with next-generation sequencing (NGS) results, PEAKS enables comprehensive immunopeptidome profiling—overcoming the limitation of single-technology approaches and ensuring accurate identification of MHC-bound peptides for cancer immunotherapy research.
#基于DIA的高效蛋白質組學工作流程 Stream-lined Proteomics Proteomics Workflow with DIA譜圖庫搜索與序列庫搜索是DIA數據分析的兩大核心方法。借助優化后的從頭測序技術,PEAKS DIA 數據分析可實現蛋白質組的深度覆蓋。
Spectral library search and library-free search are the two major approaches for DIA data analysis. Enhanced by de novo sequencing, PEAKS DIA data analysis provides you deep coverage.
#利用 PEAKS 從 DIA 數據中挖掘肽組與蛋白質組“暗物質” Uncover dark peptidome/proteome from DIA data with PEAKS
除譜圖庫搜索與直接數據庫搜索外,我們提出一種新型計算方法,可直接從復雜 DIA圖譜中鑒定肽序列變異體與新肽段,同時嚴格控制假發現率(FDR)。
In addition to library search and direct database search, we present a novel computational method to identify peptide sequence variants and novel peptides directly and solely from complex DIA spectra while rigorously controlling the false discovery rate.
#結合Intact,Top-Down與Bottom-Up技術的深度Proteoform分析 Deep Proteoform Profiling with Intact, Top-Down and Bottom-Up通過整合Intact、Top-Down(TD),與Bottom-Up(BU)技術,PEAKS可實現全面的Proteoform表征(包括亞型、翻譯后修飾組合),這一突破解決了單一方法工具無法充分解析蛋白質組復雜性的難題。
By integrating intact protein analysis, Top-Down (TD), and Bottom-Up (BU) approaches, PEAKS enables comprehensive proteoform characterization (including isoforms, PTM combinations)—a breakthrough for understanding proteome complexity that single-method tools cannot achieve.
#PEAKS GlycanFinder 實操演示 — 具有結構分辨率的糖譜學分析 PEAKS GlycanFinder Walkthrough - Glycan Profiling with Structural與只能檢測糖基是否存在的通用工具不同,GlycanFinder 可實現糖鏈結構解析(如連接方式、分支結構),解決了在糖蛋白組學研究中表征復雜糖鏈結構的關鍵挑戰。
Unlike generic tools that only detect glycan presence, GlycanFinder provides structural-level resolution of glycans (e.g., linkage, branching), solving the key challenge of characterizing complex glycan structures in glycoproteomics research.
#基于云服務器的大規模隊列蛋白質組學擴展分析 Cloud-based scalable data analysis of large cohort proteomics.
PEAKS Online 云平臺可實現大規模隊列項目的自動化分析,支持深度蛋白質組學與蛋白質基因組學分析,并能滿足發現型與靶向型質譜技術的分析通量需求。
PEAKS Online enables the automation of large-scale cohort project with in-depth proteomics and proteogenomics analyses and throughput of discovery and targeted mass-spectrometry-based technologies.
#蛋白質從頭測序 Protein de novo Sequencing


- Horticulturae為植物生命分析儀團隊開辦專題直播
- 免費講座邀請:東樂膜片鉗技術系列講座第十四期
- 南模生物原位腫瘤模型構建回歸專場活動精彩回顧
- 普瑞邦邀您參加理化檢測與真菌毒素測定交流會
- 點成直播:微流控技術驅動藥物發現與篩選的革新之路
- 進科馳安第19場BMG多功能酶標儀線上答疑會邀請函
- 10x網絡研討會:賦能臨床研究,單細胞測序入門大講堂
- 瑞沃德23周年慶典直播來襲,千元紅包面試直通等你來
- MCE全球云講堂&直播:靶向KRAS突變+LNP精準遞送解析
- 美谷分子儀器會議邀請:2025成都酶標儀用戶交流會
- 隱智科儀亮相寧波大型科研儀器維修維護發展研討會
- 講座:單細胞空間多組學探索微生物組與免疫互作機制
- 優寧維免費講座直播邀請:NK細胞的亞群與功能
- 免費講座邀請:東樂膜片鉗技術系列講座第十三期
- 紫金花論壇第九期預告:基因組學賦能心血管精準醫學
- BSI精彩亮相第24屆人類蛋白質組組織(HUPO)世界大會
- BSI邀您參加HUPO 2025,探究蛋白質組學技術最新進展
- 康昱盛邀您參加2025 Lhasa中國用戶會
- 講座:分子生成平臺Reinvent的原理與藥物AI設計功能
- PEAK蛋白質組學和生物藥專題午餐會精彩回顧
- PEAKS邀您相約蛋白質組學和生物藥專題午餐會
- 講座邀約:基于BBrowserX的肺纖維化機制探究
- PEAKS 培訓課:AI模型在高通量蛋白質組學中的應用
- 講座:Zeneth 10.1在化合物強降解途徑預測中的應用
- 第八屆PEAKS蛋白質譜數據分析培訓班日程安排
- 在線講座:深入探討新一代農藥分子設計計算解決方案
- BSI與您相約CNCP-2025,共探計算蛋白質組學前沿
- 活動報名:第八屆PEAKS蛋白質譜數據分析培訓班
- BSI邀您參加全國糖生物學會議暨全國糖化學會議
- 在線講座:BSI全新質譜軟件產品ProteoformX 介紹



