LVV體內遞送平臺技術格局、策略優劣勢和市場定位分析
文章來源公眾號:VTALK 作者:VTALK

第 1 部分:技術格局分析
1.1平臺對比矩陣
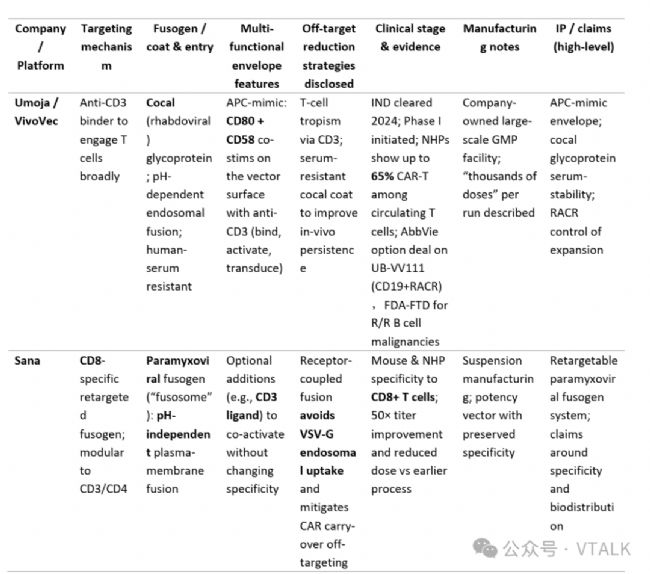
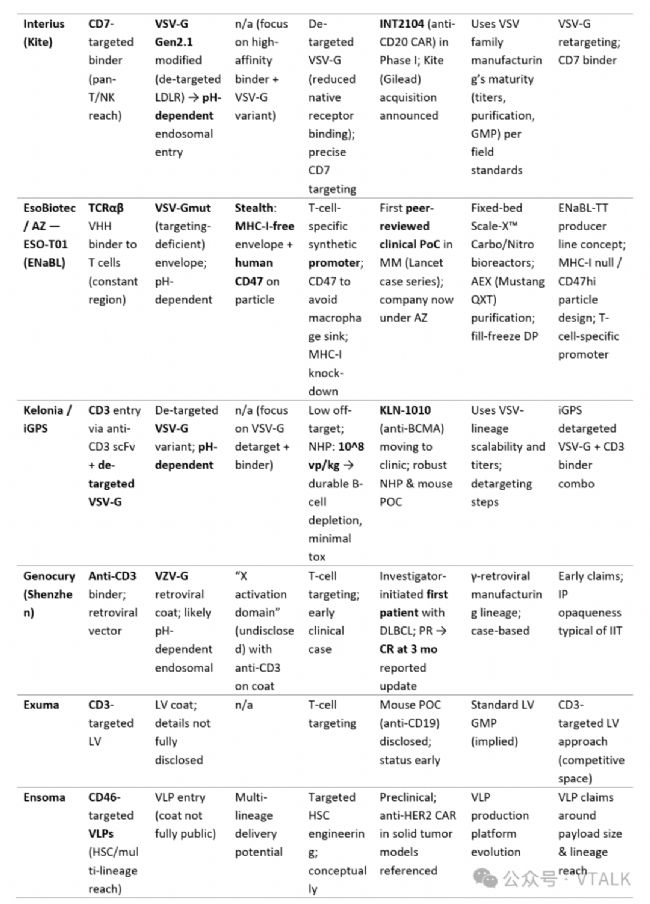
注:update to 2025。
Kelonia Therapeutics 正在開發一種創新的體內基因編輯系統 (iGPS) 平臺,用于直接在患者體內生成 CAR-T 細胞。 iGPS 平臺是通過復雜的病毒載體工程開發的:
·去靶向天然受體:對 VSV-G(水皰性口炎病毒糖蛋白)包膜進行修飾,以消除與其天然受體 LDL-R(低密度脂蛋白受體)的結合
·T 細胞特異性靶向:摻入抗 CD3 單鏈可變片段 (scFv) 以為 T 細胞提供特異性趨向性
·優化過程:篩選了多個 VSV-G 變體,以確定那些:
·最佳抑制 LDL-R 結合
·保持有效的 T 細胞轉導能力
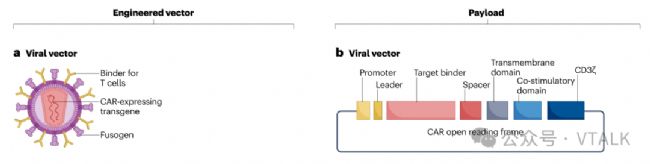
1.2 技術準備評估
·臨床驗證:ESO-T01 (BCMA) — 《柳葉刀》上第一個經過同行評審的體內 CAR-T 病例系列 (RRMM);ENaBL顆粒設計(MHC-I–/CD47+/TCRαβ VHH;T細胞特異性啟動子)在臨床補充劑中公開。
·臨床(I 期活性/啟動)階段:Umoja (CD19+RACR) 和 Interius (INT2104,CD7 靶向抗 CD20)——均已進入人體臨床研究。Kite 宣布有意收購 Interius; 艾伯維選擇了 Umoja 的 UB-VV111進行開發合作,UB-VV111于日前獲得FDA的FTD(fast track designation)。
·臨床前階段:Sana — 具有 NHP 特異性的 CD8 靶向副粘病毒融合體系統(Paramyxoviral),與早期構建的fusogen相比,滴度更高,能夠減少劑量; Kelonia — 初步展現的 NHP 藥理數據和計劃于 2025 年公布的去靶向 VSV-G CD3 管線。
·早期臨床前:Exuma、Ensoma (VLP 多譜系體內工程)和其他進入者。
第二部分:策略優劣勢分析
2.1 靶向策略分析(CD3、CD4、CD8、CD7、TCRαβ)
·CD3
o優點: 廣泛的泛-T攝取(CD4/CD8),強大的激活協同作用(例如,Umoja的CD3 + CD80 / CD58 APC模擬物);在臨床前/大型動物環境中的穩健轉導。
o缺點: 如果激活太強,更廣泛的趨向性會增加細胞因子激增的風險;如果沒有護欄,則有可能轉導 T 細胞腫瘤中的惡性 T 細胞。
o治療指標: 有利于快速 T 細胞擴增;使用 VivoVec 觀察到循環 T 細胞的 NHP 峰值高達 ~65% CAR-T。
o定位: 較多玩家的競爭(Umoja、Kelonia、Genocury、Exuma)——通過APC 模擬與隱身功能和安全導軌進行區分。
·CD4
o優點: 輔助性 T 細胞偏倚;在某些情況下可以避免惡性 CD8+ 克隆的轉導。
o缺點:CD4+ T 細胞惡性腫瘤的風險;輔助偏倚可能會降低直接細胞毒性分數。
o證據: 副粘病毒 NiV 重定向框架允許在臨床前工作中進行 CD4 特異性轉導。
·CD8
o優點:選擇性遞送至細胞毒性T細胞;Sana CD8 fusogen的臨床前/NHP數據顯示出很強的特異性和淋巴器官靶向性;可避免在T細胞惡性腫瘤中將CAR遞送至CD4+惡性細胞。
o缺點:無輔助T細胞支持,可能需要共同刺激或細胞因子支持才能持續存在。
o定位: 對B細胞癌具有很強的擬合性,具有安全優勢;利用副粘病毒 pH值不依賴的 進入優勢(第2.2節)。
·CD7
o優點:廣泛的 T/NK 覆蓋范圍 (Interius),支持多效應器 CAR 種群。去靶向的 VSV-G 變體 (Gen2.1) 增強持久性并減少意外的 LDLR 結合。
o缺點: 正常 T/NK 中 CD7 抗原表達增加了復雜性;離體潛在的自相殘殺(When a CAR targets a T-cell antigen (e.g., CD7, CD5, TRBC1/2), engineered T cells kill themselves or each other during manufacturing because they express the same antigen they’re meant to attack. Result: poor cell yields, unstable products, or the need for edits/PEBLs/knockdowns to remove)不適用于此,但生物學仍然相關;需要仔細的安全門控。
·TCRαβ(恒定區)
o優點: 靶向常規 αβ T 細胞,無論子集如何;與 MHC-I–/CD47+ 隱形涂層和 T 細胞特異性 啟動子 (ESO-T01) 配對以限制非 T 表達。
o缺點: Pan-T 與 CD3 一樣到達;在很大程度上依賴 啟動子特異性和 包膜隱蔽性來防止脫靶轉導和先天清除。
o臨床信號:通過該設計實現的第一個經過同行評審的人體療效/安全性信號 (Esobiotech)(RRMM)。


2.2 Fusogen進入機制(pH 依賴性 VSV 譜系與 pH 依賴性副粘病毒)
·特異性和脫靶
o副粘病毒(NiV/麻疹衍生):質膜融合與受體結合偶聯,→體內 VSV-G 家族可見的更高的靶向特異性和減少的非特異性內體攝取。
oVSV 譜系(VSV-G / cocal 變體): 高滴度和成熟的 GMP,但 pH 依賴性 內體融合可以允許 非特異性攝取; 緩解措施包括 去靶向 VSV-G(通過引入多個具有氨基酸取代的 VSV-G 變體,例如 Interius Gen2.1)和血清耐藥 cocal(Umoja Biopharma 的 VivoVec 平臺 使用 Cocal 病毒融合糖蛋白 而不是傳統的包膜蛋白)。
·CMC挑戰
oVSV 譜系:通常滴度最高,規模穩健,純化成熟;有吸引力的 COG。
o副粘病毒:歷史上較低的滴度;Sana 報告稱,滴度/工藝進步 50×,可在保持特異性的同時減少劑量。
·臨床應用特征
oVSV 系列現在利用對 GMP 的熟悉度,有多個已經進入臨床開發階段的LVV in vivo CAR T產品(Umoja、Interius)。
o副粘病毒 CD8 fusogen 平臺具有 NHP 特異性和生物分布優勢(報告肝臟信號降低),支持進一步開發。
·創新潛力
o副粘病毒系統對于受體重定向具有高度模塊化;VSV 譜系可以去靶向工程化處理(Kelonia、Interius)結合。
2.3 多功能Envelop工程
·APC 模擬 (Umoja):載體顯示抗 CD3 + CD80/CD58,在一個顆粒中提供結合 + 激活 + 轉導 — 加速 T 細胞原位啟動,但提出了細胞因子管理考慮因素。
·免疫隱身(EsoBiotec):MHC-I-包膜加CD47逃避吞噬作用,降低清除率/先天攝取;與T細胞特異性啟動子配對抑制非T表達。
·多譜系(Ensoma VLP): 靶向 CD46 的 VLP 旨在 HSC 產生多譜系效應子;CAR-T 處于早期階段,但有可能擴展到 T 細胞之外。
權衡:增加的envelope功能增加了 CMC 的復雜性(分析、一致性),而不是明顯的治療益處(快速激活與隱身和持久性)。Umoja 和 EsoBiotec 說明了不同的、可防御的護城河(APC 模擬與隱形啟動子+CD47)。
2.4 脫靶效應減少策略
ü受體靶向fusogen/binding domain(CD8/CD3/CD7/TCRαβ) — 特異性的主要驅動因素。
ü副粘病毒進入(受體偶聯,非pH依賴),以避免 VSV-G 出現的內體非特異性攝取。
ü去靶向的 VSV-G + 結合劑 (Kelonia、Interius)可減少天然 LDLR 結合;保留強滴度。
ü包膜上的 CD47 (和 MHC-I 減少)以減少 吞噬作用匯 和先天攝取(ESO-T01;也普遍用于 LV)。
üT 細胞特異性啟動子(以及 2025 年摘要報告中的新型合成啟動子)以最大限度地減少 整合后的非 T 表達。
ü防止載體顆粒上的 CAR 殘留 (VSV-G 的主要脫靶風險):2025 年“第四代”LVV基因組 + TRiP + 2KO 消除了顆粒中可檢測的 CAR ,并減少了異常的 vRNA 剪接。
üCAR 殘留是指在制造過程中慢病毒 (LV) 顆粒上/中意外存在 CAR 蛋白(和/或編碼 CAR 的剪接病毒 RNA)。當這些顆粒用 VSV-G 進行假分型時,病毒粒子表面的 CAR 可以將其抗原結合在非 T 細胞上并重新靶向載體,從而驅動脫靶轉導,而不受預期的融合原趨向性的影響。這種現象已被明確標記為 VSV-G 系統的安全風險:CAR 蛋白可以在 LV 生產過程中摻入載體顆粒中,并且當與 VSV-G 融合劑結合時,已被證明可以驅動脫靶轉導。
ü重要性:向攜帶抗原的旁觀者細胞(例如 B 細胞、髓系細胞或腫瘤組織)脫靶遞送會產生可避免的插入風險,并可能混淆生物分布、效力和安全性讀數。pH 依賴性內體進入途徑(VSV-G 譜系)已經容易發生非特異性攝取;在顆粒上添加 CAR 會進一步破壞特異性。
ü2025年“第四代”修復:2KO+TRiP(帶TRAP/tbs):
ü2KO 基因組:第四代LVV基因組 (SupA2KO-LV),可滅活 MSD 驅動的剪接,消除那些編碼 CAR 的剪接 vRNA。
üTRiP系統:將TRAP結合序列(tbs)插入CAR 5′ UTR(與Kozak / ATG重疊)并在 生產者系中表達TRAP;TRAP 結合結核病并抑制生產細胞中的 CAR 翻譯,防止 CAR 蛋白出現在生產者膜上(從而出現在出芽顆粒上)。
ü使用該第四代構建物重靶 NiV-糖蛋白 LV(pH 不依賴入口)顯示出更高的 T 細胞特異性, 并 避免了 VSV-G 載體在體內看到的脫靶信號(例如,骨髓/肝臟攝取)。
·藥物聯合治療:目標:在LVV給藥時瞬時提高靶向轉導和安全性,而不影響以后的CAR-T擴增/功能。
ü兩個互補boost:
l雷帕霉素(mTORC1 抑制劑)→改善 T 細胞內的載體進入/運輸。
l達沙替尼 (Src/Lck 酪氨酸激酶抑制劑)→ 抑制 TCR 信號傳導,阻止 CD3 內化,減少急性細胞因子釋放,并可以暫時“暫停”CAR 活性。
第 3 部分:競爭格局和市場定位
3.1 臨床證據層次結構
一.ESO-T01 / AZ — 第一個經過同行評審的 MM 臨床 PoC;設計包括 MHC-I–/CD47+ 包膜和 T 細胞特異性啟動子。
二.遺傳學 — 使用VZV-G逆轉錄病毒載體和抗CD3結合劑的IIT首次患者報告(iit first-patients report, DLBCL );PR → CR 更新 3 個月。
三.Umoja — I 期活性 (CD19+RACR);強大的 NHP 經驗;艾伯維選項驗證。
四.Interius (Kite) — CD7 靶向 LV + 改良 VSV-G 的 I 期;Kite 的收購表明對平臺的信念。
五.Kelonia — 晚期臨床前,NHP B 細胞耗竭強勁,濃度為 10^8 vp/kg,毒性低; IND 就。
六.Sana — 具有 NHP CD8 靶向和特異性生物分布的高級臨床前; 制造效率提高。
七.Ensoma/Exuma — 早期臨床前(分別為VLP和CD3-LV)。
3.2 適應癥策略擬合度
·血液系統惡性腫瘤(B 細胞):跨平臺最強擬合; CD19/22,由 NHP 和早期臨床信號(Umoja、Interius、Kelonia、ESO-T01)支持的 BCMA 靶點。
·T細胞惡性腫瘤:CD8靶向 方法有助于避免轉導惡性CD4細胞;存在臨床前支持。
·實體瘤: 早期(Ensoma 抗 HER2 VLP 臨床前);TME 障礙仍然存在;可能需要裝甲/連擊。
·自身免疫: 興趣日益濃厚(例如,自身免疫的Interius路線圖);安全標準高;啟動子/包膜特異性至關重要。
3.3 未滿足的需求和空白
·VSV 譜系系統的穩健脫靶硬化(脫靶向之外)→采用 TRiP/2KO 基因組和啟動子對照。
·肝臟和巨噬細胞 → CD47 和抗吞噬涂層;進一步的生物分布調整。
·用于適應癥定制的子集選擇性遞送(CD8 與 CD3/CD7); 雙靶點有效載荷以減輕抗原逃逸。
·實體瘤鎧甲裝配和 組合 策略(檢查點/趨化因子)仍處于早期體內形式。
第 4 部分:關鍵挑戰和風險評估
4.1 安全
·CRS/ICANS: 激活前向包膜 (APC-mimic) 需要仔細劑量和托珠單抗/標準措施(見于 NHP)。
·插入誘變: 根據綜合 LV 標準(監管預期)進行長期隨訪。
·長期的靶向效應: CD19/BCMA 計劃預計會出現 B 細胞發育不全(根據需要使用 Ig 替代療法進行管理)。
·免疫原性和再給藥: 包膜/結合劑免疫原性和抗載體免疫需求計劃(副粘病毒與 VSV 譜系貿易)。
4.2 功效
·轉導變異性:副粘病毒受體偶聯進入提高了 特異性; 混合包膜 NiV(75% 重靶向 + 25% 非靶向)可提高 T 細胞進入和效率。
·擴展和持久性: VivoVec 在 NHP 中顯示出強勁的峰值和二次擴展;RACR 提供藥物控制。
·抗原逃逸: 考慮LVV cargo中的早期雙抗原(例如 CD19+CD22)設計。
·TME(實體瘤): 挑戰較大
4.3 制造與可擴展性
·滴度/產量: VSV 譜系最高;副粘病毒滴度 改善 (Sana)。
·血清穩定性和配方:Cocal 涂層支持直接輸液; 隱身 功能。
·GMP 復雜性: 多域包絡和工程生產線 (ENaBL-TT) 增加了分析/控制需求。
·規模:Umoja已經進行規模化生產的準備,從自體細胞治療到真正現貨型治療是質的飛躍