藥效動力學(PD)的作用機制及主要信號轉導方式
文章來源公眾號:生物藥筆記 作者:麋鹿先生
"Pharmaco-" 這個詞來源于希臘語的 "pharmakon" —— 意思是藥物,"-dynamics" 是指與力的變化相關。所以藥效動力學(Pharmacodynamics,PD),就是研究藥物引起作用強度變化的科學。它很關心藥物效應什么時候開始、強度有多大、能持續(xù)多久這些問題,當然也關心這些效應的大小跟藥物在它作用點,作用部位那里的濃度的關系。
這里需要提一句,雖然有些書或者論文會區(qū)別使用藥物效應“effect”和藥物反應“response”,但這個區(qū)分方法目前還沒有普遍統(tǒng)一。通常情況下“效應”即“反應”。
一、藥物與受體的相互作用
1.1 作用原理
通常,藥物反應最開始的一步,是藥物分子和組織中一個大分子上的特定結合點之間發(fā)生了作用。這個大分子,就是所謂的受體(receptor)。藥物和受體相結合的時候,它會導致受體自身構象變化,這個構象變化會發(fā)出一個刺激信號,這個信號再啟動一些生物化學或者生理學上的反應最終表現(xiàn)出來(見下圖)。
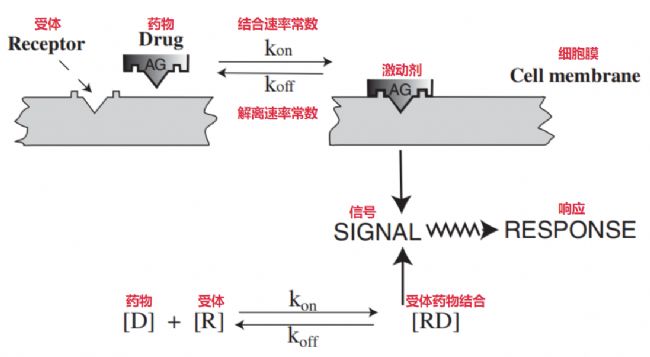 藥物-受體相互作用示意圖[1]
藥物-受體相互作用示意圖[1]
必須了解一點,絕大多數(shù)的受體超過95%其實是蛋白質構成的。但確實存在某些例外情況,比如用來做癌癥化療的一些藥物,烷化劑,它們的受體就是DNA。藥物跟受體之間的結合是靠化學鍵的,而這種結合在大部分情況下是可逆的,這個過程符合質量作用(mass action)定律的理論(見上圖)。所以在作用位點,藥物結合上受體之后,結合的藥物跟游離的藥物之間就形成一種動態(tài)的平衡。當游離的藥物最后被身體清除掉,離開了作用位點之后,結合的藥物就會從受體上解離下來,這個時候藥物引起的效應就會逐漸消失。
1.2 另一種作用
然而有少數(shù)藥物情況不同,它們會和受體形成那種基本斷不開的化學鍵結合 —— 也就是不可逆的共價鍵。比如大家都知道的阿司匹林(aspirin),它阻止血小板聚集是靠在血小板里抑制了血栓素生成。它的作用機理是跟環(huán)氧化酶發(fā)生共價結合 —— 這酶是合成血栓素的關鍵角色 —— 這種結合占據(jù)了酶的關鍵的催化區(qū)域,導致催化活性消失。結果就是,哪怕阿司匹林本身都從作用點清干凈了,吃過一次藥,那么阻止血小板的效果還是會維持著,一直到身體自己重新合成出足夠的新的環(huán)氧化酶分子出來、恢復了血栓素的生產(chǎn)才會消失。
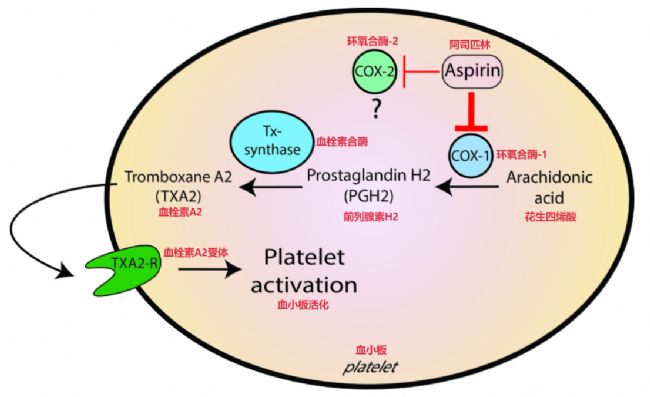 阿司匹林的作用示意圖[2]
阿司匹林的作用示意圖[2]
1.3 藥物效應關鍵
藥物能不能有效作用于受體,它的活性大小,程度取決于藥物本身和受體在化學結構上的各種特征。所以,藥物分子結構上哪怕只是一點非常微小的改變,也很可能讓它活性大大降低,甚至完全沒了活性。比如有些藥物分子像手一樣分成不同的空間構型我們叫異構體,像是R型和S型的,它們的受體能精準地分辨出這些不同。通常,其中一種構型比另一種有顯著更強的活性。華法林就是典型例子:它的 S 型異構體具有的抗凝活性大約是 R 型的 2 到 5 倍那么強。
我們得認識到受體并不是專為外源藥物存在的,所有身體里天然存在的有生物活性的物質,那些我們稱為內源性物質(endogenous compounds)或天然配體(natural ligands)的物質,像神經(jīng)遞質和激素,它們也都是作用的受體。這些天然配體和受體的相互作用,本來就是調控身體各種生理生化過程的基本方式。大多數(shù)時候藥物只是利用這一點,通過作用在對應的受體上來扮演角色,它或者是模仿內源性配體的功能(激動劑,Agonist),或者就是阻擋、干擾內源性配體的功能(拮抗劑,Antagonist)。比如:
天然的神經(jīng)遞質腎上腺素是通過激動支氣管平滑肌上的β₂腎上腺素受體來擴張支氣管的;藥物沙丁胺醇也是通過激動同一個受體來達到擴張支氣管的效果。
神經(jīng)遞質乙酰膽堿需要結合到突觸后膜上的煙堿型受體來傳遞神經(jīng)信號;像尼古丁就能模仿乙酰膽堿結合并激活這個同樣的受體,引發(fā)動作電位。
但需要特別注意一點,并非所有藥物都必須通過受體來起效。有些藥物,它們的作用機制核心在于改變機體的物理或化學環(huán)境。經(jīng)典的抗酸藥像碳酸鈣就是,它通過中和反應或者說緩沖作用來降低胃里的酸度;滲透性瀉藥聚乙二醇,則利用分子量大帶來的高滲透壓,阻止大腸吸收腸腔里的水分,這樣就產(chǎn)生了導瀉效果。
二、受體結合后的效應
藥物最終的目的是要引起細胞內的相應效應。但問題是,細胞膜那層親脂的結構,對于大多數(shù)藥物還有不少內源信號分子來說都是個物理阻礙。這就導致絕大多數(shù)受體其實是鑲嵌在細胞膜上工作的。藥物結合上膜受體后產(chǎn)生的那個最初的刺激信號,必須要傳送到細胞內部才能最終引發(fā)效應。傳輸初始刺激這個過程,被稱作“偶聯(lián)”或者更常用的“信號轉導”,通常涉及一系列按順序發(fā)生的反應步驟級聯(lián)反應,在這個級聯(lián)里面原始信號有可能被放大不少,也有可能會被弱化一些。
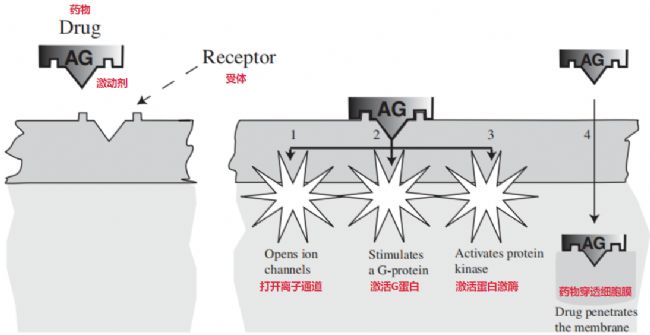 藥物受體相互作用如何引起細胞內事件的圖示[1]
藥物受體相互作用如何引起細胞內事件的圖示[1]
幾種主要的信號轉導方式簡述如下:
- 直接操控離子通道開閉:藥物結合受體后能直接讓跨膜離子通道打開或者關起來,通過改變細胞里面特定離子濃度,像鈉鉀鈣這些離子,就直接傳遞了信號。如乙酰膽堿結合煙堿受體打開鈉通道,讓鈉離子進去觸發(fā)動作電位。
- 通過G蛋白介導信號:這是最常見的一種方式了。膜上藥物-受體結合之后會觸發(fā)結合在膜里面的胞質側,觸發(fā)G蛋白活化。被激活的G蛋白接著就啟動一系列的下游事件,最終才引發(fā)生物學效應。被激活的G蛋白可以干好幾樣事:它能激活或者抑制某些酶的活性,或者直接去開關離子通道。它通常是通過改變一些重要第二信使物質的濃度來實現(xiàn)目的—— 關鍵的第二信使包括有環(huán)磷酸腺苷(cAMP)、鈣離子(Ca²⁺)還有像磷酸肌醇的衍生物比如三磷酸肌醇(IP3)和二酰甘油(DAG)這些。這些第二信使接下來再去激活比如蛋白激酶這些下游響應,通過更復雜的步驟把反應繼續(xù)傳下去。去甲腎上腺素就是這種方式。
- 激活受體相關的酶活性:藥物結合受體還能觸發(fā)受體自身關聯(lián)的酶的活性,比如一些具有酪氨酸激酶功能的結構域。被激活的激酶會把關鍵的大分子物質通過磷酸化修飾來傳遞信號,被磷酸化的目標往往是受體自身分子的一部分或者跟受體綁定的其他重要蛋白。像胰島素和多種肽類的生長因子主要就是用這種激酶關聯(lián)的方式把信號傳下去的。
還得指出的就是,有些藥物靠它們強的脂溶性能直接穿過細胞膜進去。還有一些就需要依賴于特定的轉運蛋白我們叫攝取轉運體,把它們運輸進細胞里面。一旦藥物進入細胞里面,它們就能直接跟細胞內的受體發(fā)生相互作用。典型的例子是類固醇類的激素,比如糖皮質激素、性激素、甲狀腺激素。當然,像降低膽固醇的他汀類也就是HMG-CoA還原酶抑制劑,還有治療糖尿病的二甲雙胍,它們主要在肝細胞內部起作用。這倆藥都需要靠著各自的攝取轉運體蛋白幫忙把它們運送到細胞里面作用點去才行。
三、激動劑、拮抗劑與濃度-效應關系
3.1 激動劑與濃度-效應的關系
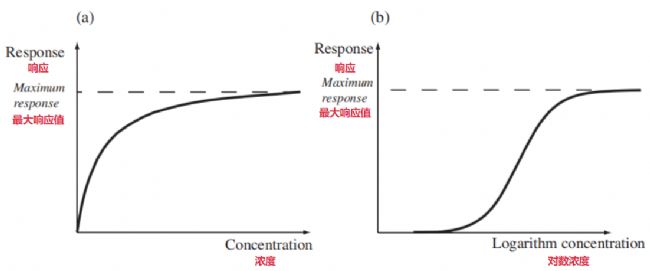
那些能模仿內源性配體去結合受體并成功將其激活的藥物,我們歸類為激動劑。下圖(a)很清楚地展示了在受體位點這里,激動劑濃度和藥物效應強度之間那個典型的關系曲線:藥物濃度增加的時候,效應強度也跟著提升。
- 在藥物濃度比較低的那個區(qū)間,效應強度和濃度基本是直線上升的關系,就是成正比。
- 隨著濃度繼續(xù)升高,再增加同樣多的濃度所獲得的效應增長量會逐漸變小。
- 濃度達到足夠高、非常高之后,效應強度就來到一個平臺期,這時候再加濃度,效應強度也基本上不動了。
 藥物濃度的響應曲線[1]
藥物濃度的響應曲線[1]
這種最終會飽和的動力學特點根本上是因為效應產(chǎn)生過程存在最大的容量限制 —— 最典型的限制因素就是這個組織里總的受體數(shù)量是有限的。在濃度比較低的時候,空著的游離受體很多,增加一點濃度就能多結合一些受體從而按比例地多產(chǎn)生一點效應。隨著濃度不斷上去,受體被占據(jù)的比例越來越大,可結合的空閑受體就越來越少,于是相同的濃度增量所能引起的效應增量也就越來越小,曲線就開始慢慢變平緩了。等到所有受體都被藥物分子給飽和占據(jù)了的時候,這個系統(tǒng)能達到的最大效應Emax就出現(xiàn)了。為了在同一個圖上能夠展示更寬濃度范圍的反應情況,特別是低濃度區(qū),人們通常把橫坐標換成對數(shù)坐標——也就是半對數(shù)坐標圖 —— 這樣畫出來的關系曲線就變成一條典型的S型曲線,見上圖(b)。有時候我們會觀察到,有些激動劑它并不需要占據(jù)全部受體就能把這個系統(tǒng)本可以達到的最大反應Emax給激發(fā)出來。這說明該系統(tǒng)存在我們說的“備用受體”現(xiàn)象。怎么證明呢?可以做個實驗,想辦法破壞一部分受體,或者把它們不可逆地封閉掉。如果做完這個操作后,給激動劑藥發(fā)現(xiàn)它還能產(chǎn)生跟原來一樣的那個最大的Emax反應,那就能驗證備用受體的存在了。藥物分子和受體一旦結合上,這個結合事件能夠被轉換成有效的原始刺激信號的效率高低,主要依賴于兩方面:
- 作用位點那里的受體數(shù)量或者說密度,另外就是這個藥物的內在效能(intrinsic efficacy)—— 內在效能意思就是每一個被藥物占據(jù)的受體單位平均能產(chǎn)生多大強度的刺激信號。
- 另一方面,在某個特定的藥物濃度下能夠達到什么樣的刺激強度,也還要看這個藥物對受體的吸引力大小 —— 也就是所謂的親和力(Affinity)。親和力高的藥物,在濃度比較低的時候就能達到可觀的結合比例從而引發(fā)反應。藥物的親和力呢,就是決定一個藥物效價強度的關鍵因素之一。
3.2 拮抗劑與濃度-效應的關系
拮抗劑是另外一類藥物,它們也能結合受體,問題是它們結合了卻不會激活受體,基本不會引發(fā)有效的刺激信號。它們主要靠占據(jù)著那個結合位點不放,把激動劑想結合過來的路給堵死了,這樣激動劑不能起到作用,如下圖。
 拮抗劑作用的示意圖[1]
拮抗劑作用的示意圖[1]
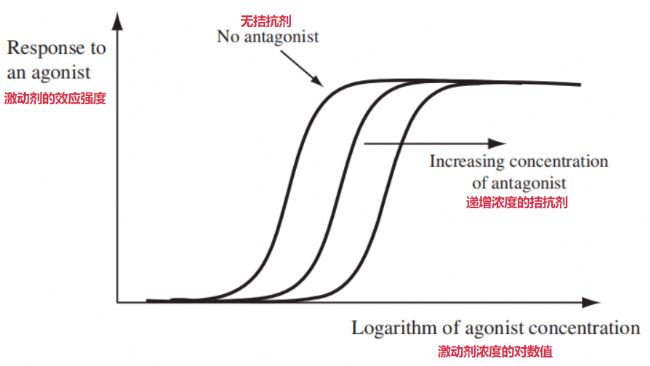
拮抗劑對激動劑的影響最直接的表現(xiàn)就是,它把激動劑那個濃度-效應曲線往右平移了—— 意思是現(xiàn)在需要更高的激動劑濃度才能產(chǎn)生原來某個程度的效應(如下圖)。如果用的拮抗劑量很大濃度很高,高到它幾乎把受體都占滿了,那么就算給高濃度的激動劑也可能不會再產(chǎn)生效應。
 激動劑在無拮抗劑和存在拮抗劑濃度增加情況下的響應曲線與對數(shù)濃度的關系[1]
激動劑在無拮抗劑和存在拮抗劑濃度增加情況下的響應曲線與對數(shù)濃度的關系[1]3.3 部分激動劑與濃度-效應的關系
另外還有一類有點特殊的藥,叫做部分激動劑(partial agonists)。它們能結合并且一定程度上激活受體,能產(chǎn)生部分效應,但這個激活能力不強。即使?jié)舛群芨咭呀?jīng)把所有的受體都占據(jù)了,這部分激動劑所能達到的最大效應Emax也要低于完全激動劑所能達到的水平,如下圖。
 完全激動劑和部分激動劑的響應曲線與對數(shù)濃度的關系[1]
完全激動劑和部分激動劑的響應曲線與對數(shù)濃度的關系[1]有意思的是部分激動劑它還有種對抗完全激動劑的能力 —— 就是當部分激動劑濃度比較高、在系統(tǒng)中占據(jù)相當部分受體的時候,它能把完全激動劑所能表現(xiàn)出來的最大效果給限制住、將其限制在它部分激動劑自己能達到的那個較低的Emax水平上。在臨床實踐中就有利用這個特性的例子。像有些β受體阻滯劑比如吲哚洛爾其實也是部分激動劑,還有像μ阿片受體部分激動劑丁丙諾啡,它們可以用作整個系統(tǒng)活動的一個緩沖器或者說是調節(jié)器。丁丙諾啡相對嗎啡的一個重要的安全優(yōu)勢,就是因為它對呼吸抑制這個危險作用有更低的一個效應上限,也就是它最大的呼吸抑制比嗎啡輕多了,這被認為是一個更安全的選擇。