DNA損傷與修復的調控機制解析
一、DNA 損傷
1.外源性損傷
外源性因素被視作引發 DNA 突變,進而誘發癌癥的關鍵力量。物理誘變劑首當其沖,其中太陽輻射中的紫外線(波長 200 - 300 納米)極具代表性。它會促使 DNA 鏈上相鄰的嘧啶堿基(胞嘧啶與胸腺嘧啶)形成共價交聯,嚴重干擾 DNA 的正常結構與功能。電離輻射(如 x 射線)則通過在細胞內催生自由基,產生高活性的氧物質(ROS),最終導致 DNA 雙螺旋結構出現單鏈或雙鏈斷裂,如同在精密的遺傳藍圖上撕開了一道道口子。
化學誘變劑也不遑多讓,它們能夠將烷基共價連接到 DNA 堿基上。以氮芥化合物為例,這類物質可使 DNA 堿基甲基化或乙基化。此外,原本化學性質惰性的致癌物原,經代謝轉化為高活性致癌物后,能與 DNA 反應生成 DNA 加合物,像苯并 [a] 芘,在細胞色素 P450 酶的介導下,經兩次氧化反應生成苯并 [a] 芘二醇環氧化物(BPDE),這種強致癌代謝物能與 DNA 共價結合,改變 DNA 的化學組成,極大地增加了基因突變的風險。
2.內源性損傷
內源性的代謝與生化反應同樣是 DNA 損傷的重要源頭。水解反應能部分甚至完全切斷核苷酸堿基與 DNA 鏈的連接。在哺乳動物細胞中,每天約發生 10,000 次脫嘌呤事件,即嘌呤堿基(腺嘌呤或鳥嘌呤)與脫氧核糖磷酸鏈間的化學鍵自發斷裂。脫嘧啶現象(胸腺嘧啶或胞嘧啶失去嘧啶基)也時有發生,只是速率相較脫嘌呤低 20 至 100 倍。
細胞內還存在脫氨反應,腺嘌呤、鳥嘌呤和胞嘧啶環上的胺基會因此丟失,分別轉變為次黃嘌呤、黃嘌呤和尿嘧啶。倘若尿嘧啶堿基在后續 DNA 復制時未被修正,極有可能被誤讀為胸腺嘧啶,從而引發 C→T 點突變。DNA 甲基化作為一種特殊的烷基化形式,在細胞內與 s - 腺苷蛋氨酸(SAM)反應后發生。在哺乳動物細胞中,甲基化常發生于距離鳥苷基(G) 5′的胞嘧啶堿基(C)的 5 位,形成序列 CpG。而甲基化的 5 - 甲基胞嘧啶產物自發脫氨后會產生胸腺嘧啶,由于其未被 DNA 修復酶識別為異常堿基,在 DNA 復制中就會產生 C→T 點突變。
正常代謝過程產生的 ROS 也會對 DNA 造成氧化損傷。嘌呤和嘧啶堿基均易被氧化,其中鳥嘌呤氧化為 8-oxo-7,8 - 二氫鳥嘌呤較為常見,對應的核苷酸 8-oxo - 脫氧鳥嘌呤(8-oxo-dG)能與脫氧腺苷堿基配對,而非正常的脫氧胞苷堿基。若此錯誤未被錯配修復酶察覺并糾正,后續復制的 DNA 將出現 C→A 點突變。ROS 還可能導致脫氧核糖核酸發生脫嘌呤、脫嘧啶以及單鏈或雙鏈斷裂。
在細胞周期 S 期的 DNA 復制階段,同樣危機四伏。負責復制模板 DNA 的聚合酶存在一定錯誤率,可能會摻入與模板 DNA 不匹配的核苷酸。化學修飾的核苷酸前體也可能被聚合酶錯誤地整合到新生成的 DNA 中。此外,當復制富含重復核苷酸或重復序列的 DNA 片段(微衛星區)時,聚合酶容易因鏈滑移,導致子鏈中核苷酸數量異常。單鏈和雙鏈裂解也可能發生,單鏈斷裂可能源于脫氧核糖基磷酸鏈的脫氧核糖部分受損,同時也是堿基切除修復途徑中 AP - 核酸內切酶作用后的中間步驟;雙鏈裂解在細胞經過 S 期時更為常見,因為此時展開的 DNA 作為復制模板更為脆弱。

二、DNA 修復機制
1.MGMT
面對 DNA 損傷,細胞進化出了多種應對策略。o6 - 甲基鳥嘌呤 DNA 甲基轉移酶(MGMT,即 DNA 烷基轉移酶)能夠從鳥嘌呤上分離甲基和乙基加合物。值得注意的是,這并非傳統的催化(酶促)反應,而是化學計量(化學)反應,每去除一個加合物就會消耗一個 MGMT 分子。研究發現,過表達 MGMT 的細胞對癌癥的抵抗力更強,或許是因為它們能有效抵消大量的烷基化損傷。近期 Niture 等人的研究表明,使用半胱氨酸 / 谷胱甘肽增強藥物和天然抗氧化劑可提升 MGMT 的表達水平,為增強細胞對 DNA 損傷的防御能力提供了新的思路。
2.DNA 聚合酶
DNA 聚合酶,如聚合酶 -δ,具備校對活性,主要參與復制錯誤修復。一旦檢測到錯誤,這些聚合酶會暫停 DNA 復制進程,反向移除子 DNA 鏈中的核苷酸,直至錯誤核苷酸被清除,隨后重新啟動正向復制。攜帶 Pold1 基因兩個拷貝點突變的小鼠,其 DNA 聚合酶 -δ 的校對活性喪失,與野生型基因或單拷貝突變小鼠相比,上皮性癌癥的發生幾率顯著升高,這充分凸顯了 DNA 聚合酶校對活性在維持基因組穩定性方面的重要性。
3.錯配切除修復(MMR)酶
錯配切除修復(MMR)酶能夠糾正 DNA 聚合酶校對活動遺漏的復制錯誤。它們從子 DNA 中移除不正確的核苷酸,并依據 W - C 配對原則,以父 DNA 鏈為模板修復子鏈。在微衛星區域復制過程中,由于 DNA 聚合酶校對活動難以察覺該區域產生的錯誤,MMR 酶的作用就顯得尤為關鍵。此外,MMR 酶還能在一定程度上糾正由 DNA 氧化或烷基化引發的各類堿基對異常,包括含有 o6 - 甲基鳥嘌呤、8 - 氧鳥嘌呤的修飾堿基對,以及致癌物和順鉑加合物。人類錯配切除修復基因 MSH2 和 MLH1 突變與遺傳性非息肉病性結直腸癌(HNPCC)綜合征密切相關,進一步揭示了 MMR 酶在預防癌癥發生中的重要意義。
三、堿基切除修復與核苷酸切除修復
1.堿基切除修復(BER)
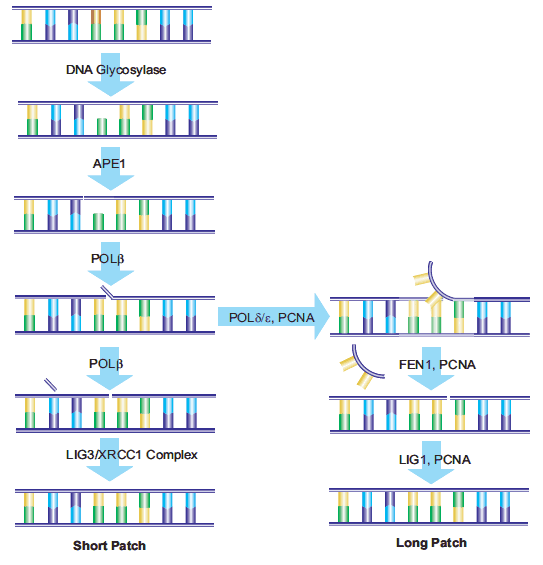
堿基切除修復(BER)主要負責切除并替換單個受損的核苷酸堿基,重點修復內源性氧化和水解導致的堿基修飾。DNA 糖基化酶率先出擊,切斷核苷酸堿基與核糖之間的連接,形成無嘌呤或無嘧啶(AP)位點。例如,8-Oxoguanine DNA 糖基化酶 I (Ogg1)能夠去除活性氧產生的堿基突變產物 7,8 - 二氫 - 8-Oxoguanine (8-oxoG),人類 OGG1 基因的多態性與肺癌、前列腺癌等多種癌癥風險相關。尿嘧啶 DNA 糖基化酶則負責切除胞嘧啶脫氨產生的尿嘧啶,有效防止后續的 C→T 點突變。n - 甲基嘌呤 DNA 糖基化酶(MPG)能去除多種修飾的嘌呤堿基。
由 BER 酶作用產生的 AP 位點,以及脫嘧啶和脫嘌呤形成的 AP 位點,可在 AP - 核酸內切酶 1 (APE1)的作用下進一步修復。APE1 在 AP 位點的 5′端裂解磷酸二酯鏈,此時 DNA 鏈一端為 3 ' - 羥基,另一端為 5 ' - 堿性脫氧核糖磷酸。接著,DNA 聚合酶 β (Polβ)依據 W - C 配對插入正確核苷酸,并利用其相關的 ap 裂解酶活性去除脫氧核糖磷酸。x 射線修復交叉互補基團 1 (XRCC1)與 DNA 連接酶 III (LIG3)形成異二聚體,作為支架蛋白,為 Polβ 提供結合位點,并將 Polβ 和 LIG3 酶聚集在修復位點。Poly(adp - 核糖)聚合酶(PARP - 1)與 XRCC1 和 Polβ 相互作用,是 BER 途徑的必要組成部分。修復的最后一步由 LIG3 完成,它將替換的核苷酸與脫氧核糖基磷酸主干連接起來,此為 “短補丁誤碼率” 途徑。
還有一種 “長補丁誤碼率” 途徑,它能替換至少 2 個核苷酸長度的核苷酸鏈,據報道修復長度可達 10 到 12 個核苷酸。長補丁 BER 需要增殖細胞核抗原(PCNA)作為重組酶的支架蛋白,可能還會調用其他 DNA 聚合酶(如 Polδ 和 Polε)產生寡核苷酸瓣,再由皮瓣內切酶 - 1 (FEN1)去除現有核苷酸序列,最后由 DNA 連接酶 I (LIG1)連接寡核苷酸,完成修復。目前,關于短補丁與長補丁誤碼率路徑選擇的具體機制仍在深入研究中。
2.核苷酸切除修復(NER)
核苷酸切除修復(NER)主要修復至少包含 2 個堿基的核苷酸鏈損傷,這類損傷往往會造成 DNA 結構扭曲。NER 既能修復單鏈斷裂,也能應對一系列外源性損傷,如大型 DNA 加合物、紫外線輻射,甚至氧化應激造成的損傷。在哺乳動物細胞中,NER 通路涉及超過 20 種蛋白質,包括 XPA、XPC - hHR23B、復制蛋白 A (RPA)、轉錄因子 TFIIH、XPB 和 XPD DNA 解旋酶、ERCC1 - xpf 和 XPG、Polδ、Polε、PCNA 和復制因子 c 等。切除修復交叉互補(ERCC1)基因的過表達與非小細胞肺癌細胞對順鉑的耐藥性相關,這反映了其增強的 DNA 修復能力。全球基因組 NER (GGR)負責修復整個基因組的損傷,而轉錄偶聯修復(TCR)作為一種特定的 NER 通路,主要在活性 RNA 聚合酶轉錄期間修復基因。
產品信息



參考文獻
[1] Hu J , Adar S . The Cartography of UV‐induced DNA Damage Formation and DNA Repair[J]. Photochemistry and Photobiology, 2017.
[2] Chatterjee N , Walker G C . Mechanisms of DNA damage, repair, and mutagenesis[J]. Environmental and Molecular Mutagenesis, 2017, 58(1):235-263.
杭州斯達特 (www.starter-bio.com)志在為全球生命科學行業提供優質的抗體、蛋白、試劑盒等產品及研發服務。依托多個開發平臺:重組兔單抗、重組鼠單抗、快速鼠單抗、重組蛋白開發平臺(E.coli,CHO,HEK293,InsectCells),已正式通過歐盟98/79/EC認證、ISO9001認證、ISO13485。