USP10去泛素化與自噬偶聯致心臟鈉通道降解并引發心律失常的機制研究
簡介:
SCN5A基因編碼Nav1.5通道,Nav1.5通道負責心肌細胞動作電位(AP)的除極和傳導,該通道維持心臟正常的電生理功能。Nav1.5的功能缺失變異會降低鈉電流密度(INa),并導致心臟傳導阻滯或Brugada綜合征等心律失常。目前對Nav1.5功能的調控機制尚未完全闡明。
基于此,武漢大學中南醫院心內科魯志兵教授團隊與華中科技大學生命科學與技術學院王擎教授團隊合作,在Cardiovascular Research發表題為“Coupling of USP10 de-ubiquitination and chaperone-mediated autophagy causes cardiac sodium channel degradation and cardiac arrhythmias”的研究論文。

本研究旨在鑒定與Nav1.5相互作用的新型蛋白質(去泛素化酶USP10),并闡明其對Nav1.5及心律失常的調控機制。
結論:
作者發現了一種新型伴侶介導的自噬(CMA)介導的通路,通過與USP10介導的Nav1.5 K430位點去泛素化作用相結合,調控Nav1.5的降解,從而導致內向鈉電流密度降低和心臟傳導異常。敲低USP10可緩解Scn5a+/−小鼠的心律失常,為治療內向鈉電流減少相關的心律失常提供了新治療策略。
研究結果:
1. 將USP10鑒定為一種新型Nav1.5-interacting 蛋白,降低心臟鈉電流密度及Nav1.5蛋白表達
為鑒定Nav1.5的新調控機制,研究人員以其胞質區域為獵物蛋白,通過GST下拉實驗篩選小鼠心臟蛋白裂解液中的互作蛋白,在−130 kDa位置鑒定出去泛素化酶USP10,質譜分析及內源性Co-IP實驗均證實二者互作。進一步通過構建USP10不同結構域表達質粒,結合Co-IP和GST下拉實驗,明確Nav1.5與USP10的USP結構域而非N端相互作用,純化蛋白實驗驗證了該互作的直接性。免疫熒光染色顯示,USP10與Nav1.5在心肌細胞中共定位。綜上,USP10通過其USP結構域與Nav1.5直接互作。
研究運用全細胞膜片鉗技術探究USP10對Nav1.5的調控:在NRCMs、HEK293/Nav1.5細胞系及hESC-CMs中,過表達USP10均顯著降低INa密度,NRCMs中失活曲線偏移而激活曲線無變化,HEK293/Nav1.5中該調控可被USP10 C424A突變消除,敲低USP10則INa密度升高。表達檢測顯示,USP10過表達降低Nav1.5蛋白總量及質膜表達,不影響Scn5a mRNA水平,敲低則提升其蛋白表達,證實USP10下調Nav1.5蛋白表達與INa密度。此外,研究發現心衰患者心臟中USP10蛋白表達較健康組顯著增加。
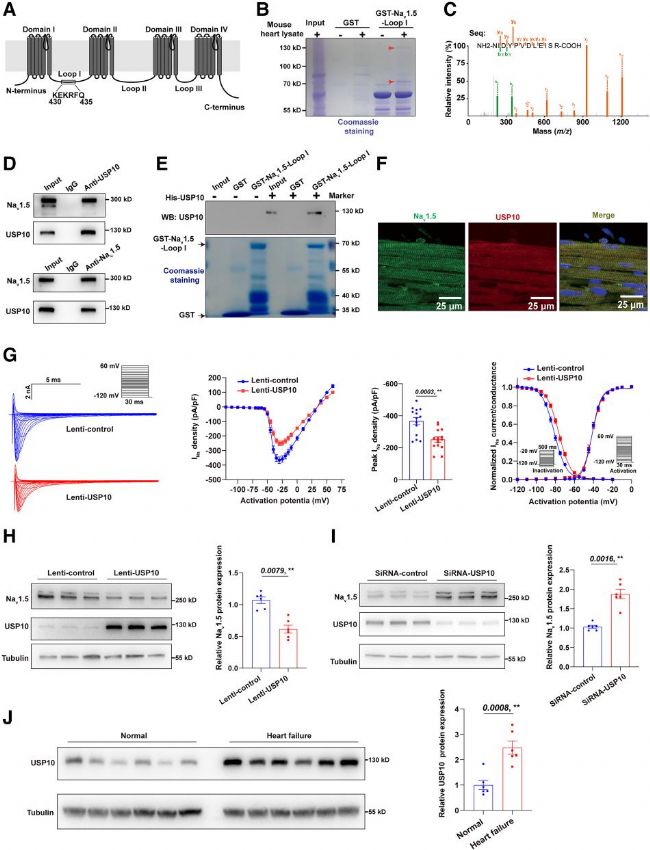
圖1. USP10被鑒定為Nav1.5的新型負調控蛋白
2. USP10過表達導致自發性心臟傳導異常,并誘導小鼠出現室性心動過速
研究通過AAV9病毒遞送系統在小鼠心臟過表達USP10,經qPCR、Western blot及免疫熒光驗證,確認USP10成功過表達且感染效率超85%。遙測心電圖顯示,過表達小鼠P波持續時間、PR及QRS間期顯著延長,出現自發性竇性停搏和房室傳導阻滯,對照組無此現象;光學標測證實其竇性及起搏模式下激活時間延長、傳導速度降低。程序性電刺激實驗表明,過表達小鼠室性心動過速(VT)誘發率高,對照組未誘發。此外,氟卡尼未誘發VT,兩組6個月均無自發性死亡,心功能無顯著差異,綜上USP10過表達導致小鼠心臟傳導阻滯并誘發VT。
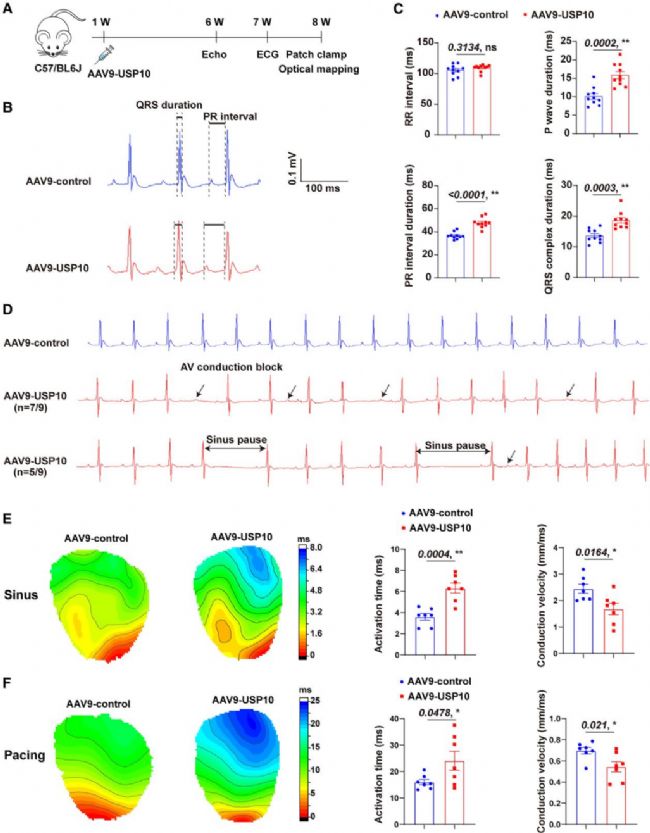
圖2. USP10過表達導致小鼠心臟傳導阻滯
3. USP10過表達改變了小鼠INa和ICa-L的密度和APD持續時間
研究分離USP10過表達的小鼠成年心室心肌細胞,檢測其離子通道電流與動作電位:全細胞電壓鉗顯示,小鼠心肌細胞INa密度顯著降低且失活閾正向偏移,晚INa無變化,ICa-L密度降低但激活/失活特性不變,Ito、IK1、Iss無組間差異;在表達人源鉀通道的HEK293細胞中,USP10也不影響人類Ito、IKr特性。動作電位檢測發現,靜息膜電位無差異,但APA、Vmax、APD90和APD50均降低,提示INa降低與傳導異常相關,ICa-L減少或致APD降低。此外,USP10過表達顯著降低小鼠心臟Nav1.5蛋白表達,而相關心律失常離子通道基因mRNA表達無差異。
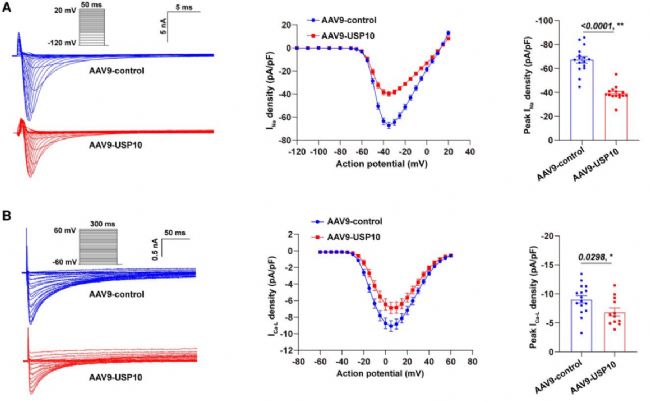
圖3. USP10過表達改變小鼠離子通道特性
4. 敲低USP10可恢復Scn5a+/−小鼠的鈉電流減少及心律失常
研究利用CRISPR-Cas9構建Scn5a+/−小鼠并注射AAV9-shRNA-USP10或對照載體,發現Scn5a+/−小鼠Nav1.5蛋白表達、INa 及 ICa-L 密度均顯著降低,Ito、IK1、Iss 電流無差異。注射AAV9-shRNA-USP10后,小鼠Nav1.5蛋白表達、INa 密度回升,ICa-L 密度恢復至野生型水平,AP參數也恢復正常。遙測心電圖顯示其傳導異常改善,室性早搏消失,證實體內敲低USP10可通過恢復INa和ICa-L密度,減輕Scn5a+/−小鼠的心臟傳導異常。
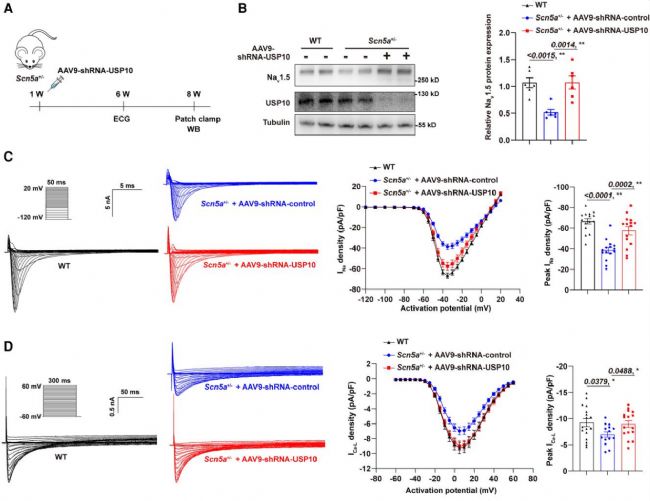
圖4. USP10基因敲除恢復了Scn5a雜合敲除小鼠中降低的INa和ICal-L
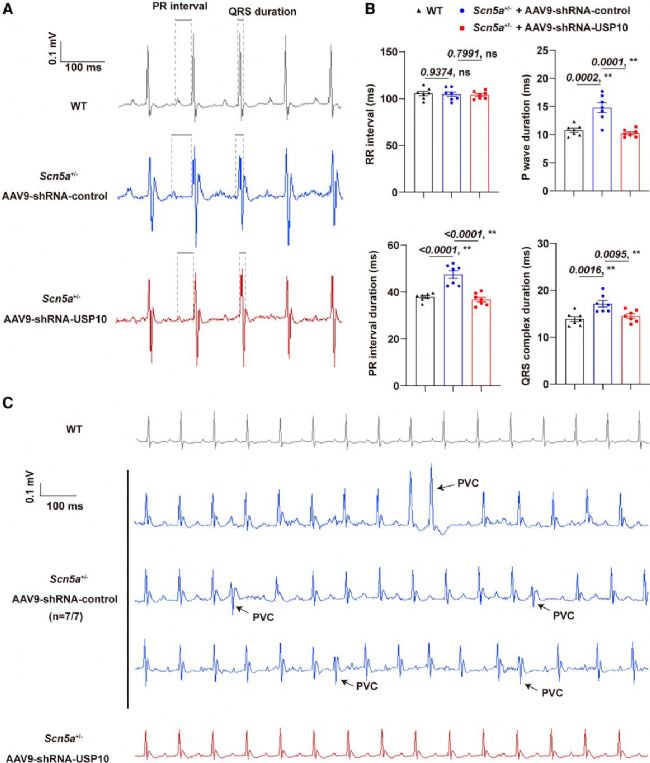
圖5. 使用shRNA敲低USP10可減輕Scn5a+/−小鼠的心律失常
5. USP10通過CMA促進Nav1.5降解,Nav1.5基序EKRFQ431—435位于環I中,參與CMA介導的Nav1.5降解
研究探究USP10降低Nav1.5蛋白表達的機制:USP10不影響Scn5a的轉錄與翻譯,而是通過降低Nav1.5蛋白穩定性促進其降解。蛋白酶體抑制劑MG132無作用,巨自噬抑制劑亦不影響,僅自噬抑制劑氯喹可阻斷該效應,且抑制CMA關鍵分子LAMP2A或HSC70能阻礙USP10功能,證實依賴CMA途徑。Co-IP顯示Nav1.5與HSC70相互作用,其環I的EKRFQ431-435基序為關鍵結合位點,該基序突變可阻斷互作、恢復Nav1.5表達及INa密度,表明Nav1.5是CMA底物,此基序介導其CMA降解。
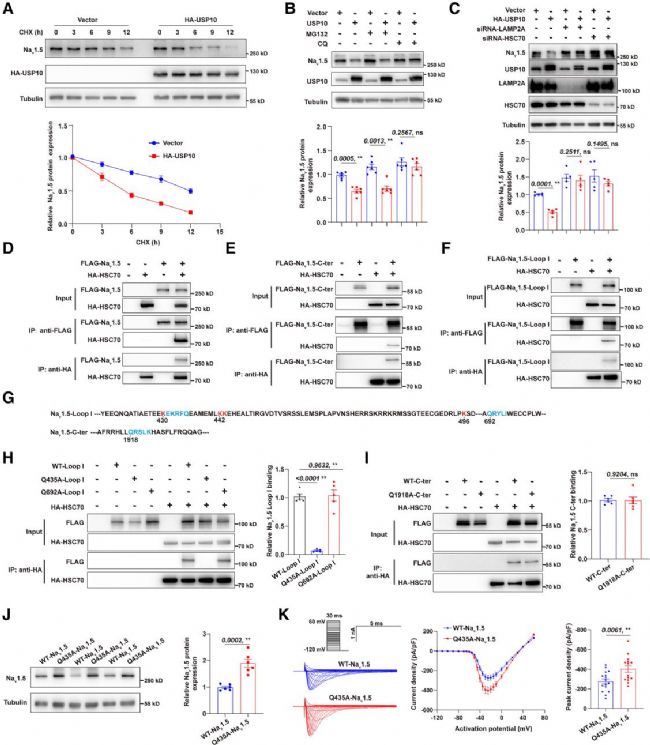
圖6. USP10通過分子伴侶介導的自噬促進Nav1.5降解
6. USP10介導的Nav1.5 K430位點去泛素化促進Nav1.5的CMA降解
研究探究USP10促進Nav1.5的CMA降解機制:因Nav1.5的 EKRFQ431-435基序緊鄰USP10結合位點K430,推測其影響 Nav1.5與HSC70的結合,實驗證實過表達USP10可顯著增強二者結合。進一步發現USP10 能降低Nav1.5泛素化水平,經突變實驗驗證,K430是其去泛素化的關鍵位點,K430R突變可阻斷該作用,還能抑制USP10對Nav1.5表達及INa密度的下調效應,揭示了去泛素化與 CMA偶聯降解Nav1.5的新機制。
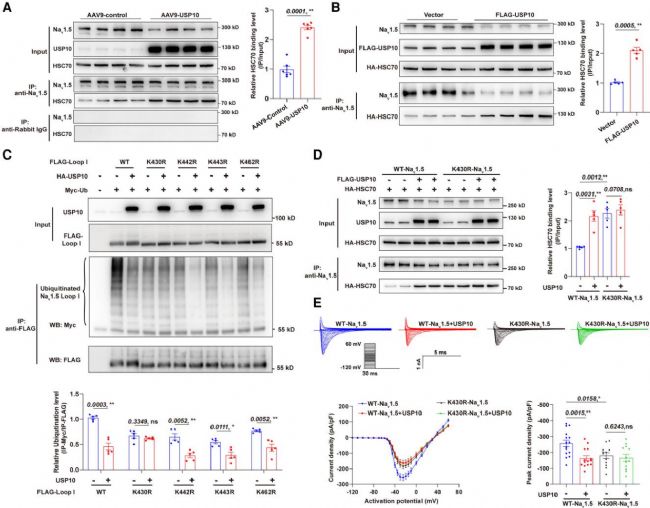
圖7. USP10對Nav1.5 K430位點的去泛素化作用增強了CMA介導的Nav1.5降解