文獻解讀:PCSK9通過LIN28A/HES5/JMY軸引發神經管畸形的研究
神經管缺陷(NTDs)是一種復雜的多基因疾病,也是影響中樞神經系統的最常見且嚴重的先天性畸形。研究已發現PCSK9可作為胎兒早期產前診斷NTD的分子標志物,但其導致在神經管發生的具體機制中仍不清楚。2025年8月,來自中國醫科大學附屬盛京醫院的袁正偉研究團隊在期刊Advanced Science(IF: 14.1)上發表了一篇題為“PCSK9 Loss-of-Function Disrupts Cellular Microfilament Network via LIN28A/HES5/JMY Axis in Neural Tube Defects”的研究文章。本研究通過將PCSK9敲除胚胎干細胞(ESC)和PCSK9 R46L點突變的誘導多能干細胞(iPSC)分別引入神經類器官(NOs)和神經祖細胞(NPCs)模型,發現PCSK9缺失通過LIN28A/HES5/JMY軸破壞細胞微絲網絡,最終導致NTDs的發生。這些發現為NTD的發病機制研究和治療策略提供了重要見解。

圖片來源:《Advanced Science》
(https://pubmed.ncbi.nlm.nih.gov/40788992/)
研究材料與方法
本研究前期從臨床檢測數據中發現PCSK9缺失與神經管畸形的發生密切相關,所以通過構建PCSK9敲除ESC和PCSK9 R46L點突變的iPSC(由賽業生物提供),分別結合三維神經類器官和二維神經祖細胞分化體系上,利用轉錄組測序篩選出關鍵分子JMY,并通過染色質免疫沉淀、熒光素酶報告基因等技術證實HES5轉錄激活JMY的表達機制。進一步通過免疫共沉淀和溶酶體抑制實驗,揭示PCSK9作為分子伴侶介導LIN28A的溶酶體降解途徑。并分別在細胞,類器官和斑馬魚模型中,通過基因敲降和過表達實驗驗證了LIN28A/HES5/JMY軸在神經管發育中的關鍵作用及表型可逆性。
技術路線
1. PCSK9 敲除 ESC、PCSK9 R46L 點突變 iPSC
2. 并行培養:
- 神經祖細胞誘導分化培養
- 神經類器官誘導分化培養
3. 轉錄組學測序分析
4. 相關機制驗證分析
研究結果
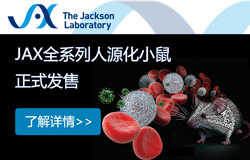
通過對人腦類器官的單細胞數據分析,發現PCSK9在胚胎發育前兩個月內高表達,隨后迅速下降,后期幾乎檢測不到。該基因主要表達于頂端放射狀膠質細胞(aRG),在部分中間祖細胞和深層投射神經元中低表達,而在其他神經元中極少表達,提示PCSK9在早期神經系統發育中起關鍵作用。
為探究其功能,技術人員構建了PCSK9基因敲除的胚胎干細胞(PCSK9⁻/⁻ ESC),通過設計靶向第二外顯子的gRNA,實現了11個堿基的缺失,導致移碼突變及提前終止密碼子,影響蛋白質功能結構域。敲除效果經Sanger測序和Western blot驗證。
進一步利用神經類器官(NO)系統比較野生型(WT)與PCSK9⁻/⁻ ESC的分化過程。結果顯示,PCSK9⁻/⁻ NOs整體形態較小,神經管(NT)結構發育不完全,周長和表面積均顯著小于WT NOs。這些結果表明,PCSK9缺失會特異性影響神經管發育并導致類器官尺寸減小。
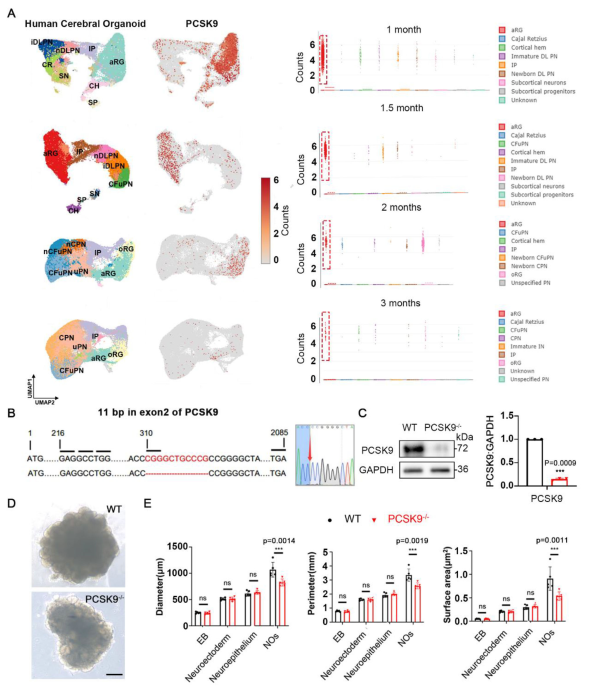
圖1 PCSK9缺失導致NOs大小和NT結構改變
(A)不同發育時間點的人腦類器官中PCSK9表達和統計結果。(B)CRISPR/Cas9介導的PCSK9位點的缺失結果。(C)WT和PCSK9-/- ESC中PCSK9的表達結果 (D)WT和PCSK9-/- NO的代表性圖像。(E)NO的直徑、周長和表面積的量化結果。[1]
對NOs橫截面的免疫熒光染色分析揭示了其內部結構特征。通過SOX2與TBR1共標記,可區分NPCs與成熟神經元。NOs在三個區域形成類神經管(NT)結構:腦室區(VZ,富含NPCs)以及腦室下區/皮質板(SVZ/CP,富含成熟神經元)。在野生型(WT)NOs中,NT結構完整,成熟神經元(TBR1⁺、CTIP2⁺)均勻分布于NPCs周圍;而PCSK9缺失導致成熟神經元減少、NPCs排列紊亂,并阻礙典型環狀NT結構的形成。神經管閉合(NTC)失敗是神經管畸形(NTDs)的重要原因,常伴隨細胞骨架異常。鬼筆環肽染色顯示,PCSK9⁻/⁻ NOs中NT區域細胞排列混亂,微絲表達增強,符合NTC障礙特征。
為進一步驗證PCSK9與NTDs的關聯,構建了PCSK9 R46L突變體及VANGL2⁻/⁻ iPSCs來源的NOs(其中PCSK9R46L和VANGL2-/- PSCs由賽業生物提供)。免疫熒光顯示,兩者均出現成熟神經元減少及環狀NT結構缺失,表型與PCSK9⁻/⁻類似。結合單細胞數據庫分析,PCSK9在發育過程中主要定位于頂端放射狀膠質細胞(aRG),屬于NPCs亞群。WT NOs中PCSK9確實富集于SOX2⁺ NPCs區域,其缺失可能引起NPCs分化異常,導致結構畸形。為進一步探究細胞表型,將WT與PCSK9⁻/⁻ ESCs誘導分化為NPCs進行培養。WT NPCs呈典型長梭形,而PCSK9⁻/⁻ NPCs則表現為多分支、排列雜亂的異常形態,通常與細胞骨架紊亂相關。微絲染色進一步證實,PCSK9⁻/⁻ NPCs結構混亂、分支增多,熒光強度增強,說明PCSK9缺失引起NPCs內細胞骨架結構異常。
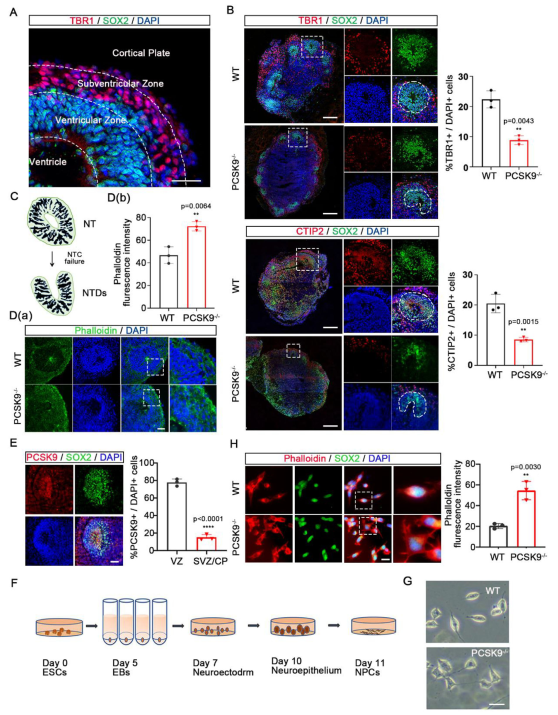
圖2 PCSK9缺失導致NPC中的細胞微絲紊亂
(A)NOs表現出三種不同的NT結構:心室、心室區 (VZ)和心室下區/皮質板SVZ/CP。(B)WT和 PCSK9-/- NO中成熟神經元的免疫熒光染色結果。正方形表示NT結構,而白色圓形或不規則形狀表示VZ區域。(C)異常NTC引起的NTD示意圖。(D)WT和 PCSK9-/- NO的NT結構中細胞微絲的免疫熒光染色結果。(E)熒光染色顯示PCSK9在WT NO的NT結構中的定位和表達結果。(F)NPC誘導培養模型圖。(G)WT 和PCSK9-/- NPC的代表性光學結果。(H)WT和PCSK9-/- NPC中細胞微絲的免疫染色結果。[1]
為解析PCSK9缺失引發NT結構異常的機制,研究人員進行了轉錄組測序分析。共鑒定出3764個差異表達基因(DEGs),其中2392個上調、1372個下調。GO富集分析顯示,這些基因在“神經系統發育”等生物學過程,以及“細胞骨架蛋白結合”等分子功能和細胞組分中顯著富集,提示PCSK9缺失主要影響神經發育和細胞骨架調控通路,與前期發現的細胞骨架異常導致NTDs相吻合。
進一步篩選出140個與細胞骨架組裝相關的關鍵DEGs,其中肌動蛋白調控因子JMY表達上調最為顯著。qPCR驗證確認其表達變化具有統計學意義。JMY已知參與Arp2/3復合體介導的微絲成核,由此提出假設:PCSK9缺失通過上調JMY,改變Arp2/3復合體活性,破壞微絲網絡,進而影響細胞形態。免疫熒光染色結果顯示,在WT神經類器官(NOs)中,JMY正常定位并沿微絲分布;而PCSK9⁻/⁻ NOs的NT結構閉合不全,JMY在細胞核與微絲中廣泛高表達,并在NT結構缺口處異常聚集,伴隨微絲信號增強。在NPCs層面,PCSK9⁻/⁻組也呈現JMY高表達及多分支紊亂形態。后續功能回復實驗證實,抑制JMY表達可有效挽救PCSK9缺失引起的表型:在PCSK9⁻/⁻ NPCs中敲低JMY能改善其多分支形態;在NOs中敲低JMY則促進環狀NT結構重建,恢復微絲正常分布,并提高成熟神經元標志物表達。綜上,這些結果表明JMY是PCSK9缺失下游的關鍵效應分子,其失調通過破壞細胞骨架重塑,導致NT結構發育異常。
圖3 PCSK9通過JMY調節NPC中微絲網絡的形成來影響NT結構
(A)GO分析中與生物過程、分子功能和細胞成分相關的項目結果。(B)細胞骨架蛋白項目中所有DEG的熱圖。(C)改變基因的qPCR驗證的統計結果。(D)細胞骨架蛋白的分子功能相關項目。(E,F)JMY和細胞微絲在NOs(E)和NPCs(F)NT結構上的表達和共定位的免疫熒光染色結果。(G)siJMY拯救實驗后WT和PCSK9-/- NPC中PCSK9和JMY蛋白水平的WB結果。(H,I)WT和PCSK9-/NPC中細胞微絲的免疫熒光染色結果。 (J,K)WT和PCSK9-/- NT結構中細胞微絲的免疫熒光染色結果。[1]
基于轉錄組數據篩選出三個可能調控JMY的轉錄因子候選,其中HES5顯示出最強的調控潛力。WB實驗顯示,在PCSK9⁻/⁻組中HES5與JMY表達同步上升,提示HES5可能正向調控JMY。通過JASPAR數據庫預測,發現HES5在JMY啟動子區存在兩個潛在結合位點(#1和#2)。ChIP實驗證實HES5在#1位點處顯著富集。熒光素酶報告基因實驗進一步表明,過表達HES5可顯著增強JMY啟動子活性。為驗證“PCSK9缺失通過HES5上調JMY”的假說,研究人員篩選出高效抑制的HES5 siRNA進行挽救實驗。結果顯示,抑制HES5可同時恢復JMY的表達水平并改善NPCs的異常形態。綜上所述,PCSK9缺失通過轉錄因子HES5正向調控JMY的表達,進而參與神經發育過程中的基因表達調控與細胞形態維持。
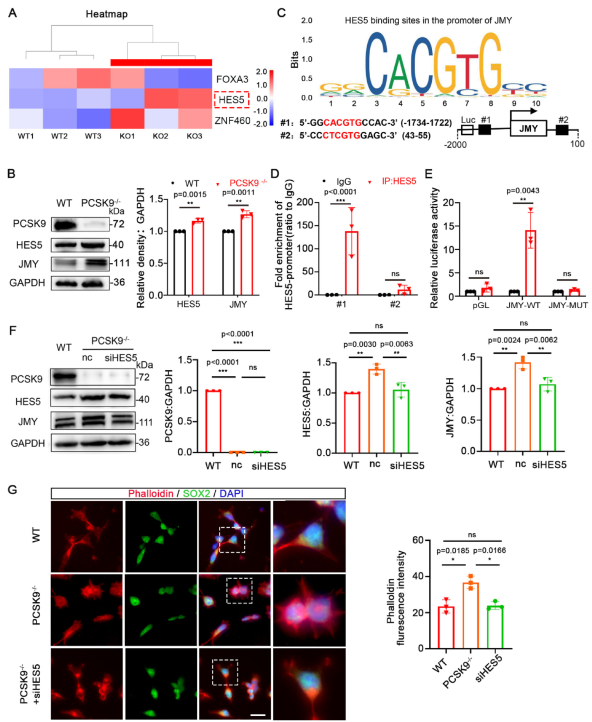
圖4 HES5作為轉錄因子調節JMY表達
(A)轉錄組數據庫中FOXA3、HES5和ZNF460表達水平的熱圖。(B)WT和PCSK9-/- NO中PCSK9、HES5 和JMY蛋白水平的WB結果。(C)HES5-JMY結合位點的預測。(D)JMY啟動子兩個位點HES5富集的ChIP-qPCR分析結果。(E)熒光素酶報告基因實驗驗證HES5表現出針對JMY的轉錄活性;(F)siHES5拯救實驗后NPC中PCSK9、HES5和JMY蛋白水平的WB結果。(G)WT和PCSK9-/- NPC中細胞微絲的免疫熒光染色結果。[1]
既往研究表明,PCSK9可作為分子伴侶,通過溶酶體途徑促進靶蛋白降解。基于此,我們推測PCSK9可能通過該途徑調控HES5水平。為探索其機制,我們通過Co-IP聯合質譜分析發現PCSK9與LIN28A存在相互作用,并在PCSK9缺失時內源性LIN28A水平顯著上升。二級質譜及結構對接模擬進一步提示兩者可能直接結合。在PCSK9⁻/⁻ NPCs中,溶酶體內LIN28A減少,而細胞質及總LIN28A水平升高,提示PCSK9可能促進LIN28A的溶酶體降解。為進一步驗證,我們分別使用CHX抑制蛋白合成和Baf-A1抑制溶酶體活性。結果顯示,在CHX處理下,PCSK9⁻/⁻組LIN28A降解減慢;而Baf-A1處理后兩組LIN28A水平無差異,說明PCSK9確實通過溶酶體途徑調控LIN28A穩定性。免疫熒光顯示PCSK9與LIN28A及溶酶體標志物LAMP1存在共定位,且PCSK9缺失導致總LIN28A升高。后續功能回復實驗表明,抑制LIN28A可改善PCSK9⁻/⁻ NPCs的多分支形態,恢復NOs中微絲表達并促進NT結構環狀重建,同時提高成熟神經元標志物水平。綜上,PCSK9缺失通過減少LIN28A的溶酶體降解,進而上調HES5與JMY表達,破壞細胞骨架結構,最終導致神經管形態發生異常。
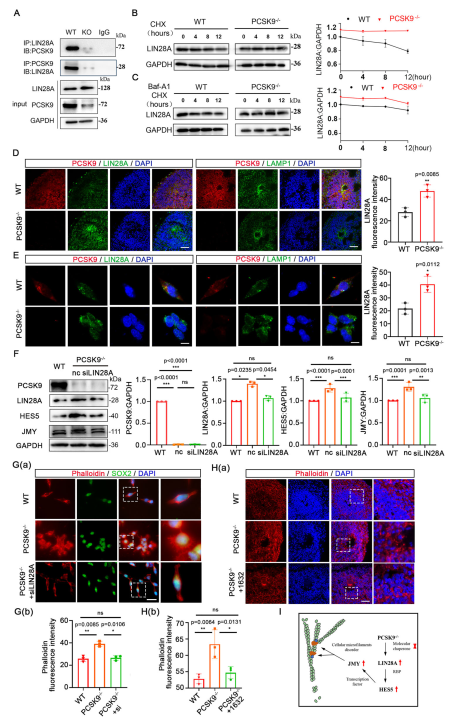
圖5 PCSK9通過溶酶體途徑促進LIN28A降解來影響NT結構
(A)PCSK9和LIN28A的Co-IP結果。(B)CHX處理后NPC的LIN28A蛋白水平的WB結果。(C)CHX和 Baf-A1處理后NPC的LIN28A蛋白水平的WB結果。 (D,E)NOs(D)和NPCs(E)NT中PCSK9、LIN28A和 LAMP1的免疫熒光結果。(F)siLIN28A救援后NPC 中PCSK9、LIN28A、HES5和JMY蛋白水平的WB結果。(G)siLIN28A救援的NPC中細胞微絲的免疫熒光染色結果。(H)抑制劑1632救援的下NT結構中的細胞微絲的免疫熒光染色結果。(I)NTD中PCSK9/LIN28A/HES5/JMY調節軸介導的細胞微絲網絡組裝改變的簡化示意圖。[1]
在斑馬魚和哺乳動物中,PCSK9均在外胚層早期發育及神經發生階段持續表達。為探究其功能,他們設計了靶向PCSK9第二外顯子的反義嗎啉寡核苷酸(MO),誘導25-bp缺失以抑制其表達。結果顯示,PCSK9-MO純合胚胎在48 hpf時出現神經管(NT)結構異常,RT-PCR證實其PCSK9轉錄水平顯著下降。盡管PCSK9-MO單獨處理僅引起23%(46/200)的神經管畸形(NTDs)發生率,但進一步實驗發現,過表達JMY mRNA可單獨誘導NTDs,并與PCSK9-MO產生協同作用:聯合處理使NTDs發生率升高至38%(76/200),并伴隨死亡率上升。組織切片顯示,PCSK9-MO斑馬魚表現為神經管縮短、管腔擴張和神經絲紊亂,聯合JMY過表達后這些表型進一步加劇。行為學分析表明,PCSK9-MO及JMY過表達均導致斑馬魚運動能力下降,表現為運動距離、速度和加速度的顯著降低,而聯合處理組上述參數下降更為明顯。綜上,斑馬魚模型中PCSK9缺失可導致神經管發育異常,而JMY過表達不僅單獨誘發NTDs,還可協同增強PCSK9缺失所引起的表型嚴重程度與發生率。
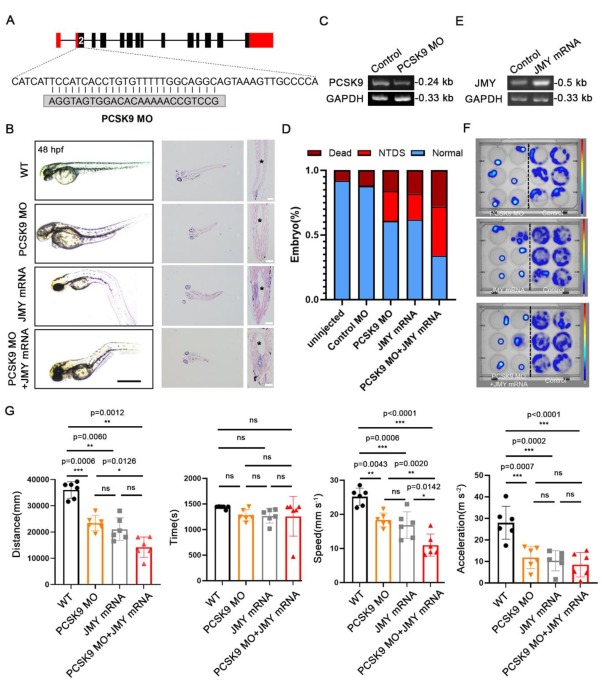
圖6 JMY加重了斑馬魚中PCSK9缺失引起的NTD
(A)針對斑馬魚第二外顯子區域設計PCSK9-MO基礎圖。(B)PCSK9-MO、JMY-mRNA及其聯合給藥對斑馬魚NT發育的影響。(C)PCSK9-MO施用后PCSK9表達的RT-PCR檢測結果。(D)PCSK9-MO、JMY-mRNA和聯合治療組中斑馬魚NTD的發病率和死亡率百分比。(E)給與JMYmRNA后JMY表達的RT-PCR檢測結果。(F)PCSK9-MO、JMY-mRNA和聯合給藥組中斑馬魚活動的熱圖像。(G)PCSK9-MO、JMY-mRNA和聯合給藥組中斑馬魚相關行為的統計分析結果。[1]
研究結論
總的來說,這項研究證明,PCSK9的功能性缺失導致神經管畸形發生是通過LIN28A/HES5/JMY信號軸導致細胞微絲網絡組裝失常的完整信號通路,為NTD的機制研究和臨床干預提供了新靶點。
參考文獻:
[1]Li X, Wang R, Luo W, et al. PCSK9 Loss-of-Function Disrupts Cellular Microfilament Network via LIN28A/HES5/JMY Axis in Neural Tube Defects. Adv Sci (Weinh). Published online August 11, 2025. doi:10.1002/advs.202504291