剪切應力對動脈粥樣硬化中血管內皮功能的影響及潛在治療策略
動脈粥樣硬化(Atherosclerosis, AS)的病理進程與血管內皮細胞(Endothelial Cells, ECs)對血流動力學的高度敏感性密切相關。臨床研究表明,AS斑塊非均勻分布于動脈系統的彎曲或分叉區域,這些部位的血流模式以振蕩剪切應力(Oscillatory Shear Stress, OSS)為主。與直動脈中穩定的層流剪切應力(Laminar Shear Stress, LSS)不同,OSS通過誘導EC表型轉化觸發促炎、促氧化應激及代謝紊亂等病理反應,導致單核細胞浸潤、脂質蓄積和斑塊形成,從而驅動AS早期病變發展。這種力學-生物學耦合效應提示,血流模式的空間異質性是決定AS區域易感性的核心因素之一。
剪切應力的力學特性通過差異激活分子通路調控EC功能穩態。LSS通過上調內皮型一氧化氮合酶(eNOS)、核因子E2相關因子2(Nrf2)和Krüppel樣因子2(KLF2)等保護性蛋白,維持EC的抗炎、抗氧化及血管舒張功能,有效抑制泡沫細胞形成和血管平滑肌細胞異常增殖。相反,OSS通過激活核因子κB(NF-κB)和活性氧(ROS)信號級聯,促進黏附分子和促炎細胞因子表達,同時破壞細胞間連接蛋白,增加內皮通透性并加速低密度脂蛋白(LDL)向內膜的滲透,最終形成惡性病理循環。
機械信號轉導機制為AS治療提供了新方向。ECs通過整合素、初級纖毛、離子通道(如Piezo1)及細胞骨架網絡將力學刺激轉化為生化信號,這一過程涉及MAPK、YAP/TAZ等多條通路。然而,現有藥物多聚焦于調控脂質代謝或炎癥因子,缺乏對力學微環境的重編程策略。基于此,四川大學華西醫院康復醫學中心與康復醫學研究所在一篇綜述中,解析剪切應力影響AS的分子機制,并探討基于力學仿生材料、基因編輯或小分子激動劑的新型治療范式。研究成果發表于“Biomedecine & pharmacotherapie”期刊題為“Effects of shear stress on vascular endothelial functions in atherosclerosis and potential therapeutic approaches”。

內皮機械傳感器響應剪切力的作用
小窩
小窩(Caveolae)作為細胞膜特殊結構,通過其核心蛋白Caveolin-1(Cav-1)參與機械應力感知與細胞保護。剪切應力可誘導內皮細胞Cav-1富集,促進小窩聚集以增強信號感知。小窩通過形態展平釋放膜面積,緩沖張力波動,維持細胞穩定性。Cav-1缺失會削弱機械信號轉導,抑制一氧化氮等舒血管介質合成,損害血管舒張與重塑功能。動物實驗證實,內皮特異性敲除Cav-1可致血流調節障礙,而重新表達則可恢復。這表明小窩通過Cav-1介導的膜張力緩沖與信號轉導雙重機制,在血流動力學調控中發揮作用。

圖1 動脈粥樣硬化進展階段
原發性纖毛
原發性纖毛是血管內皮細胞感知剪切應力的核心機械傳感器,其分布與功能具有力學依賴性。在振蕩剪切應力區域密度增加,層流區則稀疏。纖毛通過多囊蛋白復合物PC-1/2將力學刺激轉化為Ca²⁺信號并激活eNOS合成NO。PC-1缺失或PC-2異常可阻斷信號傳導,纖毛組裝障礙則加劇動脈粥樣硬化,具有保護作用。
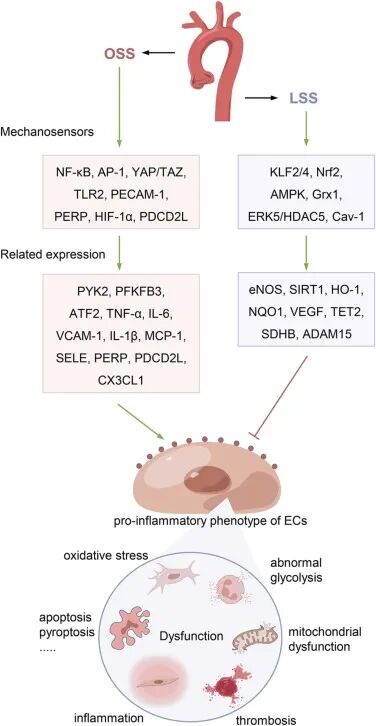
圖2 剪切應力對血管不同部位內皮細胞功能的影響。
離子通道
離子通道是內皮細胞感知剪切應力的核心元件,通過調控Ca²⁺信號調節血管功能。TRP家族成員(如TRPC1、TRPV4、TRPP2)形成復合通道響應力學刺激,介導Ca²⁺內流,觸發NO合成與血管舒張。機械敏感通道Piezo1在層流剪切應力中尤為關鍵,其激活可促進NO釋放與血管新生;內皮特異性敲除Piezo1會導致NO合成障礙、血管舒張受損及高血壓。這些通道通過時空特異性激活,將力學信號轉化為生化應答,維持血管穩態并抵抗動脈粥樣硬化進展。
連接蛋白
細胞連接蛋白通過力學信號轉導調控血管屏障與炎癥反應,PECAM-1是核心樞紐。粘附連接、緊密連接和間隙連接(如CX37/CX40)共同響應剪切應力。CX37抑制單核細胞黏附,CX40與TET1協同增強屏障完整性。PECAM-1作為機械傳感器,與Gαq/11等形成復合物傳遞信號。在AS易感區,振蕩剪切應力上調PECAM-1,促進白細胞浸潤與凋亡;其缺失在APOE⁻/⁻小鼠層流區加速斑塊,湍流區減少病變。
糖萼
糖萼是覆蓋內皮細胞的多糖蛋白復合層,通過力學化學偶聯調控屏障功能與氧化應激防御。其核心組分作為機械傳感器,層流剪切應力可增加其厚度并促進抗氧化酶表達,而糖萼缺失則削弱鈣信號與一氧化氮合成。其垂直拉伸可觸發NO釋放與鈣信號級聯,將力學刺激轉為生化應答。糖萼破壞是動脈粥樣硬化的早期事件,層流區其致密結構抑制炎癥與氧化損傷,振蕩區降解則加劇脂質沉積與單核細胞黏附。因此,靶向糖萼修復有望延緩動脈粥樣硬化進展。
剪切力及相關信號通路
eNOS信號通路
內皮細胞中不同剪切應力通過差異化調控eNOS信號通路影響動脈粥樣硬化進程。層流剪切應力通過激活PI3K/Akt和AMPK/SIRT1通路,促進eNOS磷酸化與脫乙酰化,增強NO合成,發揮抗AS作用;但同時誘導Pyk2結合并抑制eNOS,形成負反饋以維持氧化還原穩態。振蕩剪切應力則顯著降低eNOS功能,削弱內皮保護。
KLF2信號通路
轉錄因子KLF2是內皮細胞響應剪切應力的核心調控樞紐,具有抗動脈粥樣硬化作用。層流剪切應力上調KLF2,抑制糖酵解與炎癥因子,激活eNOS等血管保護蛋白,并阻斷NF-κB通路,維持血管穩態。振蕩剪切應力則降低KLF2,促進炎癥因子釋放與斑塊進展。內皮特異性KLF2缺失可加重小鼠動脈粥樣硬化病變。他汀、二甲雙胍及白藜蘆醇等藥物通過誘導KLF2表達延緩AS進程。
Nrf2信號通路
氧化應激通過活性氧失衡加劇動脈粥樣硬化,剪切應力類型決定其調控方向。層流剪切應力通過激活eNOS-NO通路及NRF2核轉位抑制ROS生成,并協同KLF2上調HO-1等抗氧化酶;振蕩剪切應力則通過NADPH氧化酶和線粒體ROS促進氧化損傷。LSS激活ERK5-KLF2/Nrf2信號軸,Nrf2缺失在某些模型中減小斑塊。
剪切力與線粒體穩態
線粒體作為內皮細胞氧化應激與炎癥的核心樞紐,既是ROS的主要來源與損傷靶點,也通過維持鈣穩態參與力學信號轉導。層流剪切應力通過提升膜電位、促進線粒體融合及增強抗氧化酶表達維持穩態;振蕩剪切應力則協同ox-LDL誘導線粒體超氧化物爆發與凋亡,驅動內皮功能障礙。
剪切力與內皮炎癥
內皮細胞的炎癥反應受剪切應力動態調控。振蕩剪切應力通過激活NF-κB和AP-1通路驅動促炎表型,上調VCAM-1、MCP-1等,促進單核細胞黏附;層流剪切應力則通過KLF2/NRF2軸抑制NF-κB/p300復合物形成,增強抗氧化防御,阻斷炎癥級聯。實驗顯示,從振蕩流切換至層流可降低促炎因子并減少白細胞浸潤。
剪切力與內皮細胞程序性細胞死亡(PCD)
機械力(如層流/振蕩剪切應力)通過內皮細胞表面機械傳感器(如離子通道、整合素)動態調節凋亡、自噬與焦亡等PCD形式。其中,層流剪切應力(LSS)抑制凋亡并激活保護性自噬,而振蕩剪切應力(OSS)則促進焦亡及異常自噬,通過炎癥級聯反應和內皮屏障破壞加速斑塊形成。靶向PCD的力學調控節點可為動脈粥樣硬化提供干預新策略。
細胞凋亡
內皮細胞凋亡在動脈粥樣硬化中具有雙重調控特征,取決于剪切應力類型。層流剪切應力通過激活KLF2依賴的VEGF/VEGFR2通路促進內皮存活,上調Adam15增強黏附,并誘導p21抑制缺氧凋亡,同時通過Grx1維持線粒體穩態。相反,振蕩剪切應力通過激活p53效應蛋白觸發凋亡,并上調PECAM-1和TLR2加劇氧化應激與炎癥。抑制mTORC2可激活自噬清除受損線粒體,阻斷氧化性凋亡。
自噬
在內皮細胞中呈現力學依賴性雙重效應:基礎自噬維持氧化還原平衡、抑制炎癥并促進NO利用,而過度自噬則引發細胞死亡與斑塊失穩。層流剪切應力通過激活SIRT1等通路增強自噬通量,抑制凋亡與衰老,減少斑塊形成;自噬缺陷則阻斷NO合成,加劇氧化應激。振蕩剪切應力作用呈參數依賴性:高強度OSS抑制自噬,加劇炎癥;低強度短時程OSS則短暫激活保護性自噬。研究顯示,自噬抑制劑僅加劇層流區斑塊進展,提示力學微環境決定自噬功能導向。
細胞焦亡
焦亡是一種新型促炎性程序性細胞死亡,通過Gasdermin D孔道形成介導細胞膜破裂。低剪切應力(如振蕩流)通過激活STAT3通路、下調TET2表達,誘導線粒體功能障礙,加劇內皮細胞焦亡及炎癥因子釋放。相比層流剪切應力,低剪切區焦亡發生率顯著升高,提示力學環境可特異性調控細胞死亡模式。褪黑激素、雌激素、紅景天苷及FGF21等通過抑制NLRP3、增強線粒體自噬或抗氧化應激,顯示抗焦亡潛力。
剪切力與內皮細胞通透性
血管內皮屏障完整性受剪切應力動態調控。層流剪切應力通過穩定黏附連接、緊密連接及間隙連接,抑制MCP-1表達并延緩糖萼降解,減少脂質滲透與炎癥浸潤;振蕩剪切應力則破壞VE-cadherin連續性,增加內皮更新與脂質沉積,加速脂肪條紋形成。糖萼作為力學敏感屏障,在層流下維持完整,振蕩流誘導其脫落,破壞內皮選擇性通透功能。
剪切力與內皮細胞及其他細胞之間的串擾
在動脈粥樣硬化進程中,剪切應力通過調控內皮細胞(EC)表型影響其與單核細胞、血管平滑肌細胞(VSMC)的病理交互。促炎性EC激活后招募單核細胞至內膜,分化為巨噬細胞并吞噬脂質形成泡沫細胞;同時EC與泡沫細胞分泌的趨化因子誘導VSMC遷移至內膜,合成細胞外基質形成纖維斑塊。這一惡性循環進一步強化EC的促炎表型,加速斑塊進展(圖3)。靶向剪切應力介導的EC-免疫/VSMC交互網絡可為干預疾病提供新思路。

圖3 剪切應力在調節內皮細胞與其他細胞間串擾中的作用
剪切力調控單核細胞對內皮細胞的粘附
振蕩剪切應力通過上調PECAM-1促進內皮凋亡與單核細胞黏附,并誘導CX3CL1表達增強THP-1細胞黏附;層流剪切應力則抑制CX3CL1激活。機械敏感通道Piezo1與TRPV4形成級聯效應,介導Ca²⁺內流破壞內皮屏障,促進單核細胞浸潤。層流應力通過穩定VE-cadherin改善屏障功能,減少單核細胞遷移。內皮細胞還可釋放miR-205/712等促炎microRNA形成正反饋環路。
剪切力在內皮功能障礙后調節平滑肌細胞表型
層流剪切應力通過維持血管平滑肌細胞收縮表型抑制其去分化與增殖,并誘導內皮細胞分泌miR-143/145促進其靜止;同時抑制促凋亡miR-126釋放,減少VSMC凋亡。振蕩剪切應力則下調收縮基因,激活PDGF-BB/TGF-β1信號,驅動VSMC炎癥與異常增殖,并促使內皮釋放miR-146a/708/451/98等抑制NF-κB通路以阻斷去分化。這些機制表明,層流應力通過力學-分子偶聯維持血管穩態,而振蕩應力破壞EC-VSMC交互網絡,誘導促炎表型并加速血管重構。
與剪切力相關的動脈粥樣硬化治療方法
研究表明,振蕩剪切應力區域PCSK9表達升高,加劇炎癥與脂質沉積,單抗藥物及siRNA基因沉默可降低風險;APOC3基因敲除有效抑制斑塊進展;層流剪切應力誘導lncRNA LASSIE及miR-146a/708/98穩定內皮屏障并抑制炎癥,而振蕩剪切應力則上調促炎miR-712/92a等。
規律運動增加層流剪切應力,促進內皮抗氧化酶表達與自噬,增強平滑肌松弛,改善血管功能并穩定斑塊。手術治療通過重構血流動力學改善預后,TAVI激活Piezo1抑制單核細胞炎癥,4D血流磁共振證實其降低主動脈渦流與振蕩剪切應力,減輕內皮損傷;PCI聯合體外反搏優化剪切應力模式穩定斑塊。
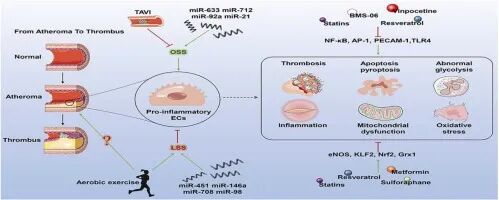
圖4 圖形摘要
總之,血流動力學剪切應力通過調控內皮細胞機械信號網絡影響動脈粥樣硬化進程。層流剪切應力激活KLF2/NRF2等保護性通路,抑制炎癥、氧化應激,維持內皮穩態;振蕩剪切應力則誘導促炎表型,促進單核細胞黏附與脂質沉積。KLF2/NRF2軸的藥物及基因療法可重構內皮功能,科學運動通過增強層流改善力學微環境。臨床干預如TAVI通過降低振蕩剪切應力減輕炎癥,但需結合計算模型優化植入參數。未來應聚焦多靶點療法、個體化運動處方及力學分子機制解析,實現精準治療突破。
參考文獻:Cheng H, Zhong W, Wang L, Zhang Q, Ma X, Wang Y, Wang S, He C, Wei Q, Fu C. Effects of shear stress on vascular endothelial functions in atherosclerosis and potential therapeutic approaches. Biomed Pharmacother. 2023 Feb;158:114198. doi: 10.1016/j.biopha.2022.114198. Epub 2023 Jan 3. PMID: 36916427.
原文鏈接:https://pubmed.ncbi.nlm.nih.gov/36916427/
圖片來源:所有圖片均來源于參考文獻
小編旨在分享、學習、交流生物科學等領域的研究進展。如有侵權或引文不當請聯系小編修正。如有任何的想法以及建議,歡迎聯系小編。感謝各位的瀏覽以及關注!進入官網www.naturethink.com或關注“Naturethink”公眾號,了解更多相關內容。
點擊了解:仿生流體剪切應力系統