ОДХВҪвЧxЈәin vivo CAR-TҜҹ·ЁЦРVivoVecВэІЎ¶ҫЯfЛНПөҪyөДСРҫҝ
ОДХВҒнФҙ№«ұҠМ–ЈәВд»ЁОўУкөДЦӘЧRҳд ЧчХЯЈәРДЧЦҜBТВ
ЧоҪьЈ¬in vivo CAR-TҜҹ·ЁҹoТЙіЙһйБЛЙъОпбtҢWоIУтЧоЦЛКЦҝЙҹбөДФ’о}ЈЎҸДҢWРg•юЧhөҪН¶ЩYХ“үҜЈ¬ҸДҝЖСРХ“ОДөҪГҪуwҲуөАЈ¬Я@№ЙҹбіұҺЧәхПҜҫнБЛХыӮҖЙъГьҝЖҢWҪзЎЈҫWҪjЙПкPУЪЯ@н—јјРgөДУ‘Х“іК¬FіцГчп@өДғЙҳO·Ц»Ҝ——Ц§іЦХЯТ•Ждһй°©°YЦОҜҹөДҪKҳOҪвӣQ·Ҫ°ёЈ¬Щ|ТЙХЯ„tҢҰЖд°ІИ«РФәНЙМҳI»ҜЗ°ҫ°іЦұЈБф‘B¶ИЎЈө«ҹoХ“ ҺЧhИзәОЈ¬in vivo CAR-TҜҹ·ЁөДСёГН°lХ№ұҫЙнҫНХfГчБЛЖдҫЮҙуөДқ“БҰәНғrЦөЎЈ
ЧчһйҝЖСР№ӨЧчХЯЈ¬ОТӮғёьРиТӘНёЯ^¬FПуҝҙұҫЩ|ЎЈЕcЖдұ»НвҪзөДРъҮМЛщёЙ”_Ј¬І»ИзмoПВРДҒнЙоИлСРЧxҺЧЖӘёЯЩ|БҝөДОД«IЈ¬ҸД»щөAҷCЦЖөҪЕRҙІ”ө“юЈ¬ҸДјјРgЖҝоiөҪОҙҒн·ҪПтЈ¬ХжХэАнҪвЯ@н—јјРgөДәЛРДЛщФЪЎЈҪсМмЈ¬ҫНЧҢОТӮғТ»Жрй_ҶўЯ@Ҳцin vivo CAR-TөДОД«IМҪЛчЦ®ВГЈЎ
Я@КЗ2024Дк8ФВ·Э°lұнФЪbloodөДТ»ЖӘОДХВЈәIn vivo CAR T-cell generation in nonhuman primates using lentiviral vectors displaying a multidomain fusion ligand. лmИ»Я@І»КЗЧоРВөДОД«IЈ¬ө«КЗVivoVecКЗUmoja Biopharmaй_°lөДВэІЎ¶ҫЯfЛНПөҪyЈ¬ЛщТФЯ@ЖӘОД«IЯҖКЗЦөөГҢWБ•Т»ПВөДЎЈХfЖрҒнЈ¬Я@КЗҪвЧxөДөЪ¶юЖӘЈ¬өЪТ»ЖӘФ”ТҠЈәОД«IйҶЧx[1]-CAR T cells produced in vivo to treat cardiac injury
01ЈәОДХВЗ°СФ
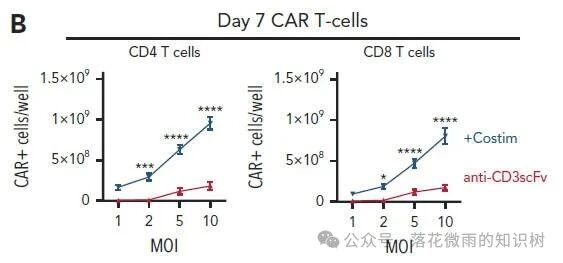
ОДХВАпЧчХЯМбөҪБЛДҝЗ°ЖХНЁCAR-TЦЖӮдөДҶ–о}ЈәТ»ӮҖЦчТӘөДҶ–о}ЦЖӮдҸНлsЈ¬РиТӘҸДГҝӮҖ»јХЯЙнЙПКХјҜҙуБҝөДTјҡ°ыЈ¬Ңўјҡ°ыЯ\ЛНөҪҢЈйTөДЦЖФмЦРРДЈ¬НЁЯ^ЯMРРуwНв»щТтҫҺЭӢәНTјҡ°ы”UФцЈ¬ҢўЖдЦЖӮдіЙЧоҪKөД®aЖ·Ј¬ЖЪйgРиТӘЯMРРQCЈ¬ЧоҪKІЕДЬҢўCAR-Tјҡ°ы®aЖ·»ШЭ”ЎЈЯ@Т»Я^іМҢ§ЦВБЛФЪіхКјTјҡ°ыКХјҜәНЧоҪK®aЖ·»ШЭ”Ц®йgҙжФЪҙуБҝөДөИҙэЖЪЎЈҙЛНвЈ¬CAR-T»ШЭ”З°ТІРиТӘЯMРРЗеБЬЈ¬¶шЗТУР•rәт°йлSЦшЭ^ҙуөД¶ҫРФЈ¬РиТӘЧЎФәЦОҜҹЎЈДҝЗ°CAR-Tјҡ°ыөДЦЖӮдәНҪoЛҺЯ^іМөДҸНлsРФЈ¬ТФј°ёЯ°әөДіЙұҫК№өГЦ»УРТ»РЎІҝ·Ц·ыәП—lјюөД»јХЯҝЙТФ«@өГCAR-Tјҡ°ыЦОҜҹЎЈ
»щУЪҙЛЈ¬ЧчХЯй_°lБЛVivoVecПөҪyЈә
»щөAЭdуwоҗРНЈәКЗөЪИэҙъЧФК§»оВэІЎ¶ҫЭdуwЎЈЯ@оҗЭdуwҪӣЯ^»щТт№ӨіМёДФмЈ¬„hіэІЎ¶ҫҸНЦЖЛщРи»щТтЈ¬ЗТҶў„УЧУ…^УтҫЯУРЧФК§»оФOУӢЈ¬ҝЙҪөөНІеИлН»ЧғпLлUЈ¬·ыәПЕRҙІ°ІИ«РФТӘЗуЎЈ
ұнГжХ№КҫөД№ҰДЬФӘјюЈә
1Ј©ЭdуwұнГжХ№Кҫҝ№CD3ҶОжңҝЙЧғЖ¬¶ОЎЈФ“scFvКЗИЛ№ӨФOУӢөДҝ№уwЖ¬¶ОЈ¬ДЬМШ®җРФҪYәПTјҡ°ыұнГжөДCD3·ЦЧУЈ¬ҢҚ¬FЭdуwҢҰTјҡ°ыөД°РПтРФҪYәПЎЈ
2Ј©ӮОРН»Ҝ°ьДӨө°°ЧЈәЭdуwІЙУГөНГЬ¶ИЦ¬ө°°ЧКЬуwЪ…ПтРФөДcocalИЪәПМЗө°°ЧЯMРРӮОРН»ҜЈәcocalМЗө°°ЧКЗДТЕЭРФҝЪСЧІЎ¶ҫGө°°Ч(VSV-G)өДҪYҳӢоҗЛЖОпЈ¬ө«ҫЯУРёьҸҠөДЙъОпҢWМШРФЈәҝ№СӘЗеңз»оДЬБҰЈ¬ПаұИӮчҪyVSV-G°ьДӨЈ¬cocalө°°ЧДЬөЦҝ№ИЛуwСaуwПөҪyөДЦРәНЧчУГЈ¬ҙ_ұЈЭdуwФЪуwғИСӯӯh•rұЈіЦ·Җ¶ЁЈ¬ұЬГвұ»ҝмЛЩЗеіэЎЈІЎ¶ҫөДcocalНЁЯ^Еcјҡ°ыұнГжөДLDLRЧR„eЈ¬ҸД¶шҢўCAR»щТтӮчЯfөҪTјҡ°ығИЎЈ
3Ј©Н¬•rұнЯ_CD80әНCD58№ІҙМјӨЕдуwЈ»
02ЈәОДХВХэОД
2.1 VVPs displaying CD80 and CD58 generate greater numbers of CAR T cells with increased in vitro and in vivo antitumor functionality.
ҝ№ФӯЯfіКјҡ°ыН¬•rПтTјҡ°ыМṩpeptide-MHCҝ№ФӯіКЯfәН№ІҙМјӨРЕМ–Ј¬ҸД¶шК№өГTјҡ°ы»о»ҜЎЈЯ@Р©РЕМ–ҢҰУЪҙ_ұЈTјҡ°ыјӨ»оЈ¬ҙЩЯM·Ц»ҜЈ¬«@өГР§‘Ә№ҰДЬәНРОіЙйLЖЪУӣ‘ӣЦБкPЦШТӘЎЈCD28әНCD2Ј¬ТФј°ЛьӮғёчЧФөДЕдуwCD80әНCD58Ј¬КЗҳӢіЙTјҡ°ы-ҝ№ФӯМбіКјҡ°ыөДкPжI№ІҙМјӨРЕМ–ЎЈЧчХЯІВңyЈ¬ФЪПИЗ°Х№Кҫҝ№CD3 scFvөДVVPsЦРјУИлCD80әНCD58№ІҙМјӨЕдуwЈ¬ҢўФцҸҠЖдјӨ»оTјҡ°ыөДДЬБҰЈ¬ҸД¶ш®aЙъҫЯУРёДЙЖҝ№Д[Бц№ҰДЬөДCAR Tјҡ°ыЎЈһйБЛтһЧCЯ@Т»јЩФOЈ¬ЧчХЯҳӢҪЁБЛVVPsәН№ІұнЯ_CD80әНCD58өДVVPsЈ¬ІўФЪуwНвЕарBөДҪЎҝөИЛPBMCsЦРФu№А2·N№ІҙМјӨЕдуwөДЧчУГЈЁS1A-BЈ©ЎЈІ»Н¬УЪөдРНөДК№УГҝ№CD3/CD28ҙЕЦйҙМјӨөДуwНвЦЖӮдCAR-Tјҡ°ыөД·Ҫ°ёЈ¬VVPsЦұҪУМнјУөҪPBMCsЦРЈ¬¶шІ»РиТӘИОәОНвФҙРФҙМјӨЎЈ№ІҙМјӨ·ЦЧУөДјУИлҙуҙуФцҸҠБЛІЎ¶ҫоwБЈҪYәПTјҡ°ыөДДЬБҰЈЁS1CЈ©ЎЈ
¶шЗТ№ІҙМјӨ·ЦЧУөДјУИлТІҢ§ЦВ¶МЖЪTјҡ°ы»о»ҜФцҸҠЈ¬Я@АпКЗVVPsјУИлИэМмәуЈ¬НЁЯ^БчКҪҷzңyCD25өДұнЯ_Л®ЖҪтһЧCөДЈЁ1AЈ©ЎЈ
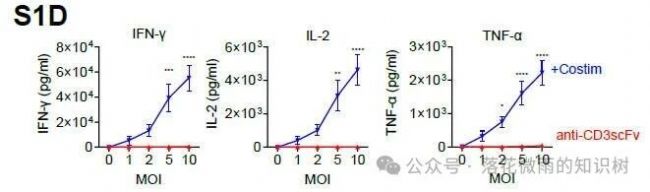
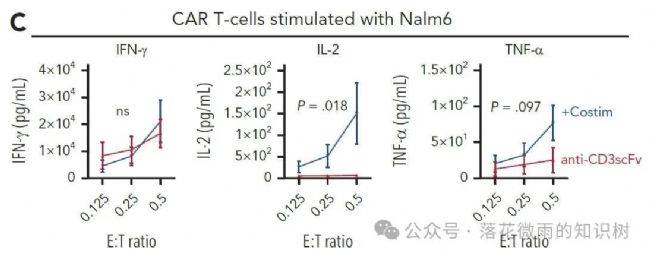
¶шЗТ№ІҙМјӨ·ЦЧУөДјУИлТІҢ§ЦВTјҡ°ыбҢ·ЕБЛёь¶аөДјҡ°ыТтЧУЈ¬°ьАЁIFNγЈ¬IL-2әНTNF-αЈЁS1DЈ©ЎЈ
І»Х“КЗ·сә¬№ІҙМјӨЕдуwөДVVPs¶јДЬ®aЙъПаЛЖұИАэөДCAR-Tјҡ°ыЈЁS1EЈ©ЎЈ
ө«КЗУР№ІҙМјӨЕдуwөДVVPsҝЙТФ®aЙъёь¶аөДCAR-Tјҡ°ыЈЁ1BЈ©ЎЈ
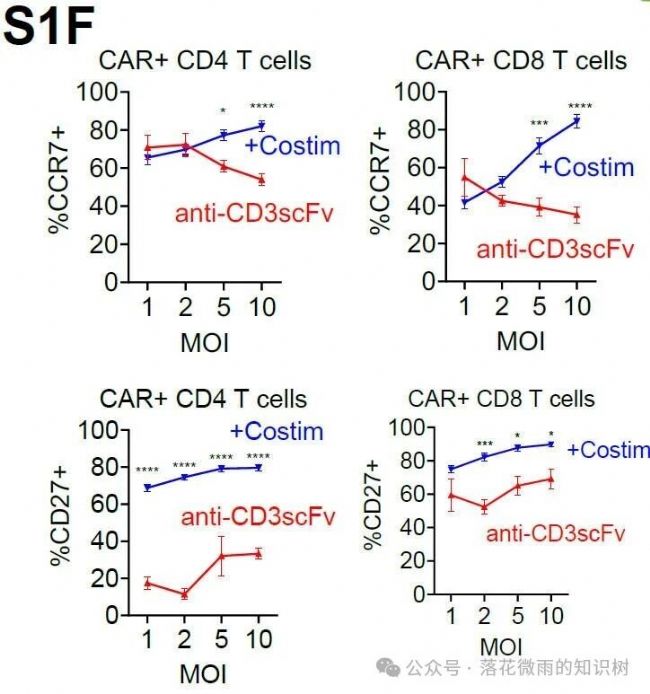
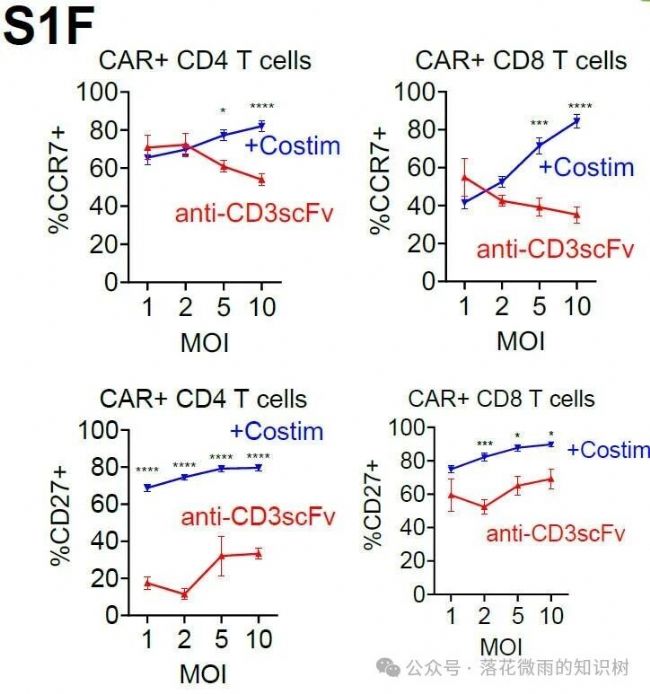
ұнРНЙПЈ¬ұнЯ_№ІҙМјӨ·ЦЧУөДVVPs®aЙъөДCAR-Tјҡ°ыұнГжұнЯ_ёь¶аөДCCR7әНCD27ЈЁS1FЈ©ЎЈЯ@ғЙӮҖmarkerКЗУӣ‘ӣРФTјҡ°ыИәПакPөДҳЛЦҫОпЈ¬ЕcуwНвCAR-Tјҡ°ыЦОҜҹөДЕRҙІ·ҙ‘ӘіКХэПакPЎЈҝӮөДҒнХfЈ¬Я@Р©”ө“юұнГчЈ¬ә¬УРCD80әНCD58№ІҙМјӨЕдуwөДVVPsұн¬FіцФцјУөДTјҡ°ыҪYәПәНјӨ»оЈ¬ҸД¶шҢ§ЦВёь¶аөДCAR-Tјҡ°ыЙъіЙЈ¬ІўҫЯУРЭ^өН·Ц»ҜөДTјҡ°ыұнРНЎЈ
ҢўұнЯ_»тІ»ұнЯ_№ІҙМјӨ·ЦЧУөДVVPs®aЙъөДCAR-Tјҡ°ы·Ц„eЕcNalm-6јҡ°ыТФІ»Н¬өДETұИ№ІЕарBЈ¬24РЎ•rәуЈ¬ҷzңyјҡ°ыТтЧУөДұнЯ_ЎЈұнЯ_№ІҙМјӨ·ЦЧУөДVVPsҪMөДCAR-Tјҡ°ыұИҶОӘҡұнЯ_ҝ№CD3 scFvөДVVPs®aЙъөДIFNγөДБҝПаЛЖЈ¬ө«IL2әНTNF-αөДЛ®ЖҪёьёЯЈЁ1CЈ©ЎЈ
ұнЯ_№ІҙМјӨ·ЦЧУөДVVPsҪMөДCAR-Tјҡ°ыЕcNalm-6јҡ°ы№ІЕарBәуЈ¬CAR-Tјҡ°ы”UФцөДТІёь¶аЎЈЯ@АпКЗУЙCellTrace dilutionҒнЕР”аөДЎЈҸДҲDЦРҝЙТФҝҙөҪЈ¬CostimҪMөДCellTraceөДҹЙ№в¶јЖ«ИхЈ¬ХfГчЯ@ӮҖҪMөДCAR-Tјҡ°ы”UФцёьҸҠЈ¬ФміЙCellTrace Dyeұ»ПЎбҢЈЁS1GЈ©ЎЈ
ҢўVivoVecЙъіЙөДҝ№CD19 CAR-Tјҡ°ыЈЁ·ЦғЙҪMЈәә¬№ІҙМјӨ·ЦЧУCD80/CD58өДVVPs vs. ғHә¬anti-CD3 scFvөДVVPsЈ©ЕcұнЯ_CD19өДД[Бцјҡ°ыЈЁNalm6Ј©№ІЕарBЎЈГҝ2-4МмТЖіэЕfЕарB»щЈ¬ЦШРВМнјУРВхrД[Бцјҡ°ыЈЁҲDЦРјэо^ұнКҫҙМјӨ•rйgьcЈ©Ј¬іЦАmұOңyД[Бцјҡ°ы”өБҝЧғ»ҜЈЁНЁЯ^ҹЙ№вҳЛУӣ¶ЁБҝЈ©ЎЈә¬№ІҙМјӨ·ЦЧУөДVVPsҪMөДCAR-Tјҡ°ыіЦАmТЦЦЖД[БцЙъйLЈ¬јҙК№ФЪ¶аҙОҙМјӨәуИФДЬУРР§ҝШЦЖД[Бцјҡ°ы”өБҝЈЁҲDЦРЛ{Й«ЗъҫҖҫSіЦөНО»Ј©ЎЈғHә¬anti-CD3 scFvөДVVPsҪMЈ¬лSҙМјӨҙО”өФцјУЈ¬Д[Бцјҡ°ыЦрқuФцЦіЈЁјtЙ«ЗъҫҖЙПЙэЈ©Ј¬ұнГчCAR-Tјҡ°ы№ҰДЬЛҘҪЯ»тіЦҫГРФІ»ЧгЈЁ1DЈ©ЎЈ
ҙЖРФNod.Cg-PrkdcscidIL2rgtm1Wjl/SzJРЎКуЈЁЯ@ӮҖРЎКуЦШ¶ИГвТЯИұПЭҢ§ЦВT/Bјҡ°ыИұК§Ј»IL2rg»щТтЗГіэИұ·ҰNKјҡ°ыЈ»MHC I/IIлpЗГіэЈ©ЎЈDay -4Ј¬НЁЯ^ ОІмoГ}ЧўЙд 2.5E5ӮҖ Nalm6јҡ°ыЎЈDay -1Ј¬УГ IVISңyБҝРЎКуД[БцШ“әЙЎЈёщ“юД[БцҙуРЎлSҷC·ЦҪMЈ¬ҙ_ұЈҪMйg»щҫҖТ»ЦВЎЈНЁЯ^ё№З»ЧўЙд2E7ӮҖPBMCsЎЈDay 0Ј¬ё№З»ЧўЙд”yҺ§CAR»щТтөД VivoVecІЎ¶ҫоwБЈЈЁ„©Бҝёщ“ю·ЦҪMХ{ХыЈ©Ј¬уw·e200 μLЈЁS1EЈ©ЎЈ
VVPҪoЛҺ4МмәуЈ¬ИЎРЎКуөДНвЦЬСӘЈ¬НЁЯ^БчКҪјҡ°ыРg¶ЁБҝ·ЦОцИЛФҙCD3+ Tјҡ°ыұнГжCD25әНCD71өДұнЯ_Л®ЖҪЈ¬Фu№АЖд»о»Ҝ о‘BЎЈ1EөДҷMЧшҳЛҙъұнөДКЗІ»Н¬VivoVecІЎ¶ҫоwБЈөДЧўЙд„©БҝЈ¬ҶОО»һйЮDҢ§ҶОО»ЈЁTUЈ©ЎЈЕcуwНвСРҫҝҪY№ыТ»ЦВЈ¬ұнЯ_Һ§УР№ІҙМјӨЕдуwөДҝ№CD3 scFvөДVVPsұИҶОӘҡұнЯ_ҝ№CD3 scFvөДVVPsХTҢ§БЛёьҸҠөДTјҡ°ы»о»ҜЈ¬ЗТіК„©БҝТАЩҮРФЎЈVVPҪoЛҺ11МмәуҷzңyНвЦЬСӘЦРCAR-Tјҡ°ыоlВКЎЈУГә¬№ІҙМјӨЕдуwөДҝ№CD3 scFvөДVVPsМҺАнөД„УОпп@Кҫіцёь¶аөДCAR-Tјҡ°ы”өБҝЈЁ1F-GЈ©ЎЈЧоәуЈ¬ҢҰД[БцШ“әЙЯMРРФu№АЈ¬К№УГ№ІҙМјӨЕдуwұнЯ_ҝ№CD3 scFvөДVVPsЦОҜҹЈ¬ФЪVivoVecІЎ¶ҫоwБЈЭ^өН„©БҝПВД[БцТЦЦЖЧчУГҫНәЬҸҠБЛЈЁ1HЈ©ЎЈ
2.2 An MDF protein comprising costimulatory molecules enhances T-cell activation and transduction.
ЧчХЯФOУӢБЛТ»·N°ьә¬¶аӮҖҪYҳӢУтөДө°°ЧЈЁMDFЈ©Јә°ьАЁCD58Ўўҝ№CD3 scFvәНCD80ҪMіЙөДҶОжңйLлДЎЈЯ@ҳУҝЙТФәҶ»ҜVVPsөДЦЖӮдЈ¬ІўЗТіЙ№ҰҢўMDFіЙ№ҰХ№КҫФЪІЎ¶ҫұнГжЈЁS2AЈ©ЎЈ
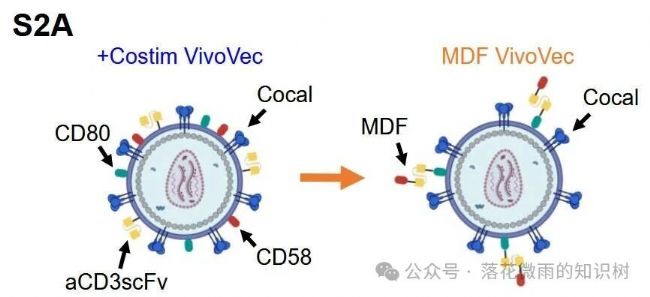
іцәхТвБПөДКЗЈ¬ЕcҶОӘҡөДҝ№CD3 scFv»тә¬УР№ІҙМјӨ·ЦЧУөДІЎ¶ҫоwБЈПаұИЈ¬MDF VVPsЕcTјҡ°ыөДҪYәПТӘёь¶аЈЁ2AЈ©Ј¬ІўҢ§ЦВёьёЯөДTјҡ°ыCD25ұнЯ_ЈЁ2BЈ©ЎЈ
ө«КЗMDF VVPsІЎ¶ҫоwБЈХTҢ§өДјҡ°ыТтЧУІўӣ]УРГчп@ФцјУЈЁS2BЈ©ЎЈ
ЦөөГЧўТвөДКЗЈ¬MDF VVPsФЪуwНвДЬёьУРР§өШ®aЙъCAR-Tјҡ°ыЈ¬УИЖдКЗФЪөНёРИҫҸН”ө•r(2C)ЎЈ
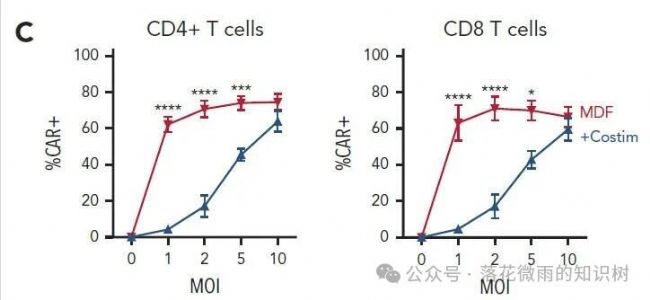
Еcә¬УР№ІҙМјӨ·ЦЧУөДІЎ¶ҫоwБЈПаұИЈ¬MDF VVPsЦЖӮдөДCAR-Tјҡ°ыұнГжұнЯ_Па®”өДCCR7әНCD27Уӣ‘ӣјҡ°ыҳЛЦҫОпЈЁS2CЈ©ЎЈ
ҪУПВҒнҫНКЗЈ¬ЕcұнЯ_№ІҙМјӨ·ЦЧУөДVVPsҪMПаұИЈ¬MDF VVP®aЙъөДҝ№CD19 CAR-Tјҡ°ыФЪЕcNalm6јҡ°ы№ІЕарB•rЈ¬CAR-Tјҡ°ы”UФцөДБҝІоІ»¶аЈЁ2EЈ©Ј¬IFNγ®aЙъБҝІоІ»¶аЈЁ2DЈ©Ј¬ө«®aЙъёь¶аөДIL-2әНTNF-αЈЁ2DЈ©Ј¬МбКҫMDF VVP®aЙъөДCAR-Tјҡ°ыҝЙДЬҫЯУРёьәГөД№ҰДЬРФЎЈФЪЯBАmҙМјӨҢҚтһЦРЈ¬MDF VVP®aЙъөДCAR-Tјҡ°ыДЬүтёьәГөШҝШЦЖNalm6Д[БцөДЙъйLЈЁ2FЈ©ЎЈ
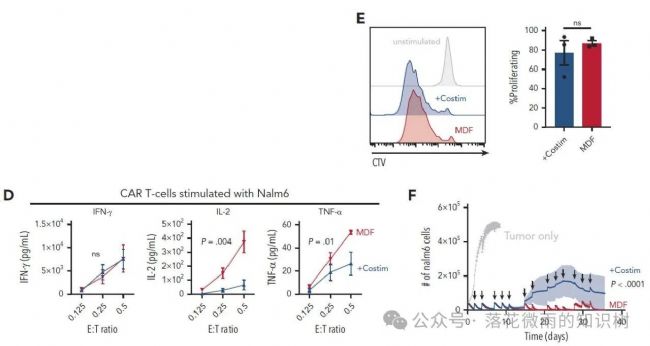
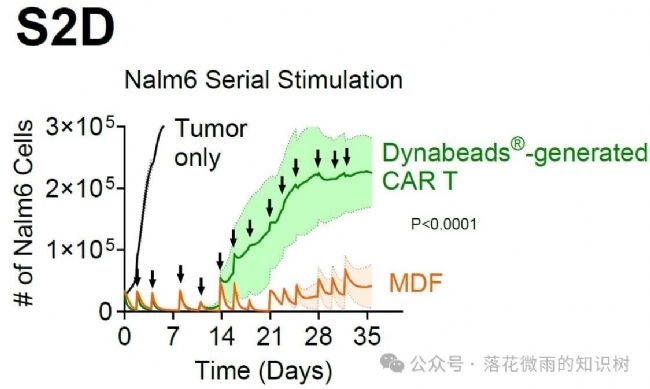
ФЪЯBАmҙМјӨҢҚтһЦРЈ¬MDF VVP®aЙъөДCAR-Tјҡ°ыЙхЦБғһУЪК№УГӮчҪyөДCD3/CD28 beadsһй»щөAөДҙМјӨ·Ҫ°ё®aЙъөДCAR-Tјҡ°ы(S2D)ЎЈ
2.3 MDF VVPs exhibit enhanced in vivo functionality in a humanized mouse leukemia model.
ЧчХЯФЩҙОК№УГNSG MHC I/II KOДЈРНтһЧCMDF VVPөДҝ№Д[БцЧчУГЎЈ VVPҪoЛҺ4МмәуЈ¬ИЎРЎКуөДНвЦЬСӘЈ¬НЁЯ^БчКҪјҡ°ыРg¶ЁБҝ·ЦОцИЛФҙCD3+ Tјҡ°ыұнГжCD25әНCD71өДұнЯ_Л®ЖҪЈ¬Фu№АЖд»о»Ҝ о‘BЎЈЕcуwНвСРҫҝҪY№ыТ»ЦВЈ¬MDF VVPsұИұнЯ_№ІҙМјӨөДVVPsХTҢ§БЛёьҸҠөДTјҡ°ы»о»ҜЈ¬ЗТіК„©БҝТАЩҮРФЈЁ3AЈ©ЎЈVVPҪoЛҺ11МмәуЈ¬ЧчХЯУ^ІмөҪғЙ·NІЎ¶ҫоwБЈФЪСӘТәЦР®aЙъБЛПаЛЖұИАэөДCAR-Tјҡ°ыЈ¬ө«MDF VVPsІЎ¶ҫоwБЈҝЙТФ®aЙъёь¶аөДCAR-Tјҡ°ы”өДҝЈЁ3BЈ©ЎЈЯ@АпЕcFigure1ПаұИЈ¬3BЦРCAR-Tјҡ°ыөД”өДҝГчп@ЙЩБЛәЬ¶аЈ¬ЧчХЯҪoіцөДҪвбҢКЗТтһйІ»Н¬donorІ»Т»ҳУөДФӯТтЎЈ
лmИ»ғЙ·NІЎ¶ҫоwБЈҫщДЬҝШЦЖД[БцЙъйLЈ¬ө«MDF VVPsФЪҝШЦЖД[БцЙъйLәНҝӮЙъҙжЖЪ·ҪГжұн¬FВФәГЎЈ
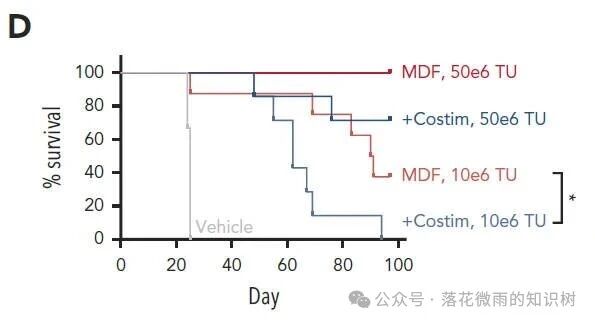
Я@ФЪ10E6ӮҖЮDҢ§ҶОО»өДЭ^өН„©Бҝ•rУИһйГчп@Ј¬MDF VVPsЦОҜҹ„УОпөДЦРО»ЙъҙжЖЪһй90.5МмЈ¬¶шұнЯ_№ІҙМјӨ·ЦЧУөДІЎ¶ҫоwБЈҪMөДЦРО»ЙъҙжЖЪһй62МмЈЁ3DЈ©ЎЈЦөөГЧўТвөДКЗЈ¬ФЪ50E6ӮҖЮDҢ§ҶОО»өДЭ^ёЯ„©БҝПВЈ¬MDF VVPМҺАнөД„УОпФЪСРҫҝЖЪйgЯ_өҪБЛ100%өДҙж»оВКЎЈЯ@Р©”ө“юұнГчЈ¬ФЪЯ@ӮҖДЈРНЦРЈ¬Х№КҫMDFө°°ЧөДVVPsПаҢҰУЪұнЯ_ҝ№CD3 scFvәНұнЯ_№ІҙМјӨЕдуwөДVVPsҫЯУРёьәГөДҝ№Д[БцР§№ыЎЈ
2.4 MDF VVPs administered intranodally induce potent and prolonged B-cell depletion in NHPs.
ИЛФҙ»ҜРЎКуДЈРНҫЯУРҫЦПЮРФЈ¬ІўІ»ДЬНкИ«ФЩ¬F„УОпГвТЯПөҪyөДНкХыіМ¶ИЈ¬МШ„eКЗНкИ«Иұ·ҰХэіЈ°lУэөДҙОјүБЬ°НҪMҝ—әНҢҰ®җ·NТЖЦІОпҝ№ЛЮЦчІЎөДТЧёРРФЎЈһйБЛҪвӣQЯ@Р©ҫЦПЮРФЈ¬ЧчХЯФЪM nemestrinaЦРй_°lБЛТ»ӮҖNHPДЈРНЈ¬ТтһйФ“Оп·NФКФSУГHIV-1СЬЙъөДВэІЎ¶ҫЭdуwЮDҢ§ЎЈФЪЯ@ӮҖДЈРНЦРЈ¬ЧчХЯК№УГБЛТ»·NNHP/ИЛҪ»Іж·ҙ‘ӘөДҝ№CD20 CARЈ¬ҙЛЗ°ТСҪӣЧCГчФЪәгәУәпуwғИК№УГуwНвЦЖФмөДCAR-Tјҡ°ыҝЙТФПыіэBјҡ°ыЎЈһйБЛК№MDFө°°ЧЯmУГУЪФ“ДЈРНЈ¬ЧчХЯҢўҝ№ИЛCD3 scFvМж“Qһйҝ№NHP CD3 scFvЎЈИЛөДCD58әНCD80ұЈБфПВҒнЈ¬ТтһйЯ@ғЙ·Nө°°ЧФЪЯ@ғЙӮҖОп·NЦРөДРтБРҫЯУРёЯ¶ИН¬ФҙРФЈЁS3AЈ©ЎЈ
Фu№АMDF VVPsөДNHPМжҙъОпҢҰM.nemestrina CD4әНCD8 Tјҡ°ыөДҪYәПЎўјӨ»оәНЮDҢ§ЯMРРуwНвтһЧCЈЁS3BЈ©ЎЈЕc°Рјҡ°ыLLC-MK2-NLO+/-CD20№ІЕарBЈ¬тһЧCCAR-Tјҡ°ыөДјҡ°ыҡўӮы№ҰДЬЈЁS3CЈ©ЎЈҝӮөДҒнХfЈ¬Я@Р©ҪY№ыұнГчЈ¬NHP MDF VVPsҪYәПЎўјӨ»оәНЮDҢ§NHP Tјҡ°ыөДДЬБҰЕcИЛMDF VVPsПа®”ЎЈ
NHPДЈРНЈәәҶСФЦ®Ј¬НЁЯ^БЬ°НҪYЧўЙдЈ¬Ңў”yҺ§ҝ№CD20 CARШ“ЭdөДNHP MDF VVPsТФІ»Н¬„©БҝЧўЙдөҪ4Ц»„УОпөДБЬ°Н№ЬПөҪyЎЈЕcмoГ}ҪoЛҺНҫҸҪПаұИЈ¬Яx“сБЬ°НҪYҪoЛҺНҫҸҪҝЙТФҢҚ¬FУРР§өДІЎ¶ҫоwБЈЕcTјҡ°ыПа»ҘЧчУГЈ¬Н¬•rңpЙЩИ«ЙнҪMҝ—ҢҰУОлxІЎ¶ҫоwБЈөДұ©В¶ЎЈ„УОпГҝЦЬИЎСӘ1-2ҙОЈ¬НЁЯ^БчКҪјҡ°ыРgҷzңyІЎ¶ҫоwБЈХTҢ§өДTјҡ°ы»о»ҜЎўCARұнЯ_әНлSәуөДBјҡ°ыәДҪЯЎЈН¬•rҢҰ„УОпЯMРРCRSәНICANSуwХчұOңyЎЈФЪЯ@ӮҖҢҚтһЦРЈ¬ЧчХЯјИӣ]УРЗеБЬЈ¬ТІӣ]УРо~НвСaідјҡ°ыТтЧУЎЈЖдЦРЈ¬1Ц»„УОп(Z20020)ҪУКЬБЛөН„©БҝөДVVPsЈЁФӯ„©БҝөДОе·ЦЦ®Т»Ј©ЎЈ
ҪoЛҺәу7МмЈ¬4Ц»„УОпЦРУР3Ц»У^ІмөҪNHP MDF VVPХTҢ§өДTјҡ°ы»о»ҜЈЁ4AЈ©ЎЈФЪЯ@3Ц»„УОпЦРЈ¬NHP MDF VVPҪoЛҺР§№ып@ЦшЈ¬ФЪөЪ10Мм®aЙъөДCAR Tјҡ°ыХјСӯӯhTјҡ°ыөДұИАэёЯЯ_65% (4B)Ј¬Па®”УЪГҝОўЙэСӘТәЦРә¬УР5963875әН11182ӮҖCAR-Tјҡ°ыЎЈөН„©БҝҪMөДZ20020, ЧчХЯӣ]УРУ^ІмөҪГчп@өДTјҡ°ы»о»ҜЈ¬CARөДұнЯ_Ј¬ТІӣ]УРУ^ІмөҪBјҡ°ыөДПыіэЎЈ
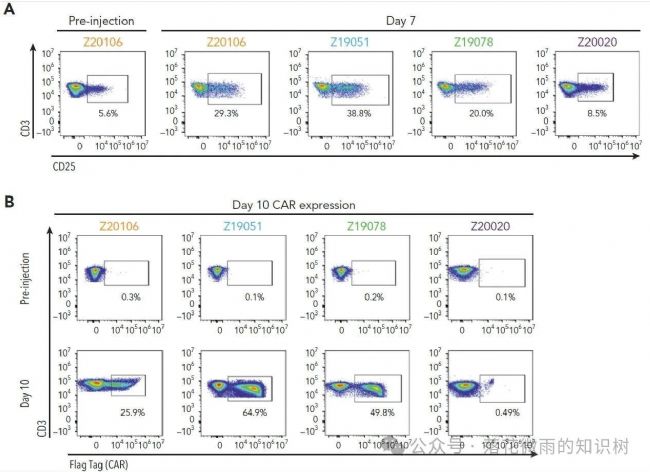
ХэИзоAЖЪөДДЗҳУЈ¬өҪөЪ7МмЈ¬ФЪЛщУРҷzңyөҪCAR-Tјҡ°ыөД„УОпЦРЈ¬СӯӯhBјҡ°ы¶јҷzңyІ»өҪБЛЎЈХfГчCAR-Tјҡ°ыҙуБҝ®aЙъЕcBјҡ°ыПыіэКЗН¬•rіц¬FөДЈЁ4C-DЈ©ЎЈПаұИЦ®ПВЈ¬ФЪ®aЙъCAR-Tјҡ°ыөД3Ц»„УОпЦРЈ¬СӯӯhBјҡ°ыөДҒGК§КЗТ»ЦВөДЈ¬ТІ·ЗіЈіЦҫГЎЈФЪЦОҜҹәуөДөЪ56Ўў63әН76МмЈ¬СӯӯhBјҡ°ыҺЧәхҷzңyІ»өҪЈЁZ19051ЙФОўІоТ»ьcЈ¬Bјҡ°ыФЪ63әН76ЯҖКЗДЬҝҙөҪТ»ьcBјҡ°ыЈ©ЎЈЦөөГЧўТвөДКЗЈ¬1Ц»„УОп( Z20106 )ФЪөЪ49Ммп@КҫіцҙуБҝөДСӘТәCAR Tјҡ°ыөДЦШРВ”UФцЈ¬ТФ‘ӘҢҰФЪСРҫҝөДөЪ42МмГчп@өДЙЩБҝBјҡ°ыөДФЩҙОіц¬FЎЈЯ@ұнГчVivoVec®aЙъөДCAR-Tјҡ°ыҫЯУРіЦҫГРФЈ¬Я@оAКҫЦшУӣ‘ӣРФCAR-Tјҡ°ыөД®aЙъЎЈЕcУӣ‘ӣРФTјҡ°ыөДРОіЙТ»ЦВЈ¬Bјҡ°ыТІФЪөЪ¶юЦ»„УОпөДөЪ43Мм( Z19078 )өДСӯӯhЦРЦШРВіц¬FЈ¬И»әуФЪөЪ49МмФЩҙОҒGК§ЎЈЦұөҪөЪ63МмЈ¬Bјҡ°ыФЪФ“„УОпЦРИФИ»ҷzңyІ»өҪЈ¬Я@ұнГчҝ№ФӯМШ®җРФУӣ‘ӣCAR-Tјҡ°ы№ҰДЬіЦАmҙжФЪЎЈ
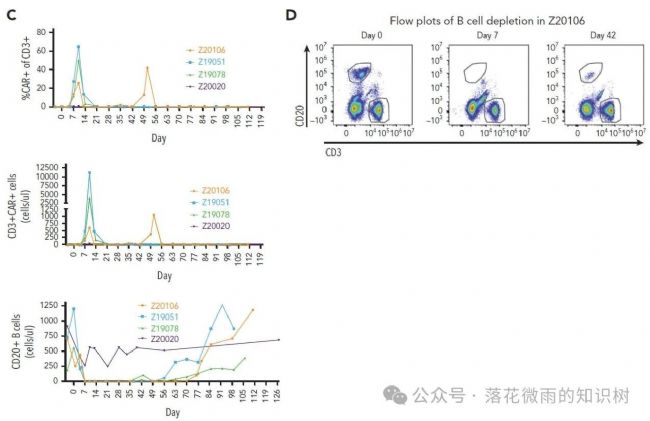
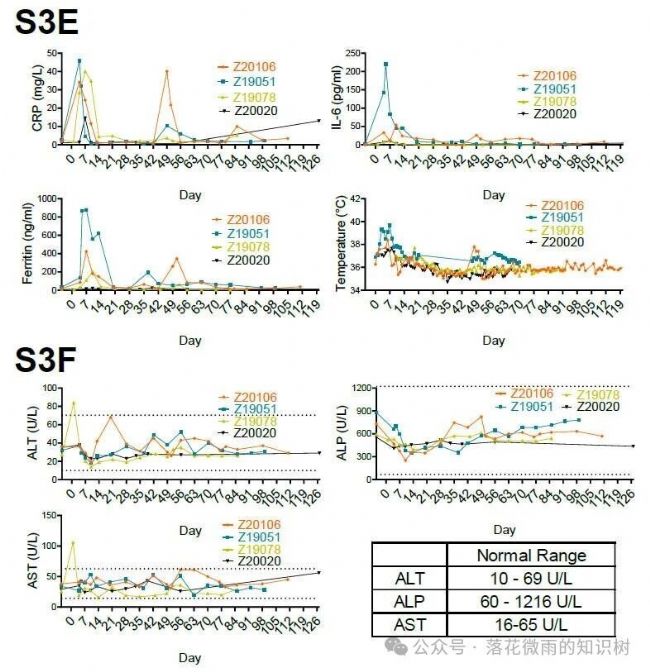
ФЪCRS·ҪГжЈ¬ЧчХЯФЪСӘТә(S3E)ЦРҷzңyөҪЈ¬CAR-Tјҡ°ы®aЙъөДЗ°І»ҫГҫН•юУ^ІмөҪC·ҙ‘Әө°°Ч(CRP)Л®ЖҪЙэёЯЈ¬Я@ұнГчЯ@КЗCAR-Tјҡ°ыҪйҢ§өДЈ¬¶шІ»КЗІЎ¶ҫоwБЈҢ§ЦВөДЎЈЯ@КЗ¶М•әөДЈ¬лSЦшCAR-Tјҡ°ыФЪөЪ14МмҸДСӘТәЦРПыК§Ј¬CRPЛ®ЖҪХэіЈ»ҜЎЈИзЙПЛщКцЈ¬1Ц»„УОпФЪөЪ49МмҪӣҡvБЛСӘТәCAR-Tјҡ°ыөД»ШЙэ(4C)Ј¬Я@ТІФЩҙО°йлSЦшCRPөД¶М•әЙэёЯЎЈIL-6Ј¬иFө°°ЧәНңШ¶ИЧсСӯоҗЛЖөДТҺВЙЎЈіэБЛCRS°Y оЈ¬ЧчХЯФЪЗ°2Ц»„УОпЦРУ^ІмөҪЭpОўөДХроқәН¶М•әөД°d°BЎЈЯ@Р©КВјюКЗ¶М•әөДЈ¬ЕcөЪ10МмУ^ІмөҪөДCAR-Tјҡ°ы”UФц·еЦөПаОЗәПЎЈҪoУиНРЦйҶОҝ№әН°ўДЗ°ЧңюЛШЦОҜҹәуХроқПыК§ЎЈЧчХЯӣ]УРУ^ІмөҪИОәОГчп@өДёОЕK¶ҫРФөДЫEПуЈ¬Еc№ИұыЮD°ұГёЈ¬№ИІЭЮD°ұГёЈ¬үAРФБЧЛбГёИФИ»ұЈіЦФЪХэіЈЛ®ЖҪЈ¬іэ1Ц»„УОпөД№ИұыЮD°ұГёәН№ИІЭЮD°ұГёФЪөЪ1Мміц¬F¶М•әЭp¶ИЙэёЯНвЈЁS3FЈ©ЎЈ
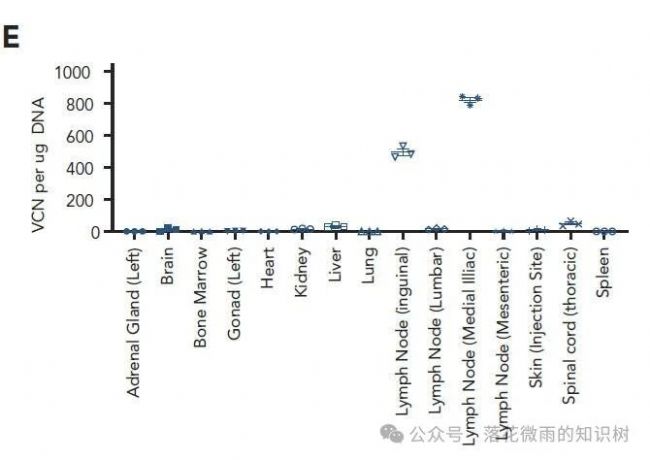
ИЎVVPЦОҜҹЧойL•rйg(139Мм)өД1Ц»„УОпЯMРРЖКҷzЈ¬20·NІ»Н¬ҪMҝ—НЁЯ^ddPCRФu№АЭdуw»щТтҪMХыәПЎЈФЪЧўЙдөДІҝО»inguinal lymph nodeәНҫoҪУЦшөДmedial iliac lymph nodeҝЙТФҷzңyөҪҝ№CD20 CARЮD»щТтЎЈө«КЗФЪЖдЛьҪMҝ—ЦРІўӣ]УРҷzңyөҪЈ¬Я@ұнГчФЪVivoVecМҺАнәуЈ¬ЮD»щТтјҡ°ыЦчТӘұ»ёфлxФЪЧўЙдәНаҸҪьөДБЬ°НҪYЦРҺЧӮҖФВЈЁ4EЈ©ЎЈ
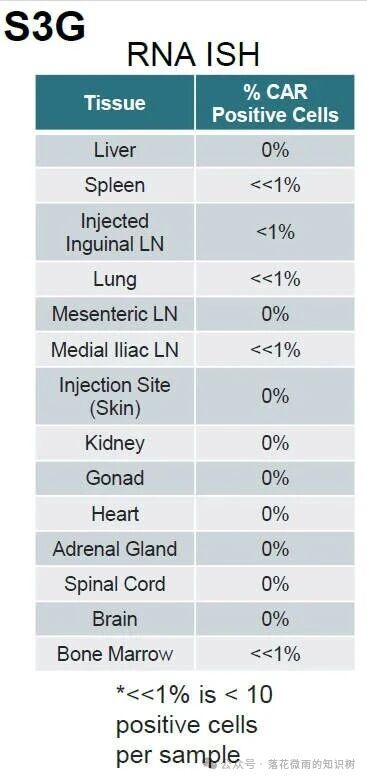
ЧчХЯНЁЯ^RNA ISHҷzңyБЛCAR»щТтөДЙъОп·ЦІјЈЁS3GЈ©ЎЈЕcddPCR”ө“юТ»ЦВЈ¬ЧчХЯФЪЧўЙдөДІҝО»inguinal lymph nodeәНҫoҪУЦшөДmedial iliac lymph nodeҝЙТФҷzңyөҪҳOЙЩ”өјҡ°ыЦРCARөДұнЯ_ЎЈЯҖФЪЖўЕKЎў№ЗЛиәН·ОЦРҷzңyөҪөНЛ®ЖҪөДРЕМ–Ј¬ЧчХЯХJһйЯ@Р©РЕМ–КЗCAR+јҡ°ыПтЯ@Р©ІҝО»ЮDЯ\өДҳЛЦҫЎЈҙЛНв„eөДҪMҝ—ғИТАИ»ҷzңyІ»өҪЎЈҝӮөДҒнХfЈ¬Я@Р©”ө“юұнГчЈ¬VivoVecНЁЯ^БЬ°НғИЧўЙдЈ¬ФЪӣ]УРЗеБЬөДЗйӣrПВЈ¬ҝЙТФФЪГвТЯҪЎИ«өДNHPЦРУРР§өШЙъіЙCAR-Tјҡ°ыЎЈЙъіЙөДCAR-Tјҡ°ы№ҰДЬҸҠҙуЈ¬Ң§ЦВBјҡ°ыЗеіэйLЯ_76МмЎЈ
03Јә·Ҫ·Ё…ўҝј
3.1 Vector productionЈә
Ңў5·NЩ|БЈН¬•rЮDИл‘ТёЎЕарBөДHEK 293Tјҡ°ыЈәgag pol(ҫҺҙaІЎ¶ҫҪYҳӢө°°ЧЈЁТВҡӨЎўГёөИЈ©); rev(…fЦъІЎ¶ҫRNAЮDЯ\іцјҡ°ыәЛ); cocal(ҫҺҙaөНГЬ¶ИЦ¬ө°°ЧКЬуwКИРФөДІЎ¶ҫ°ьДӨМЗө°°ЧЈЁМжҙъVSV-GЈ©); MDF(ҫҺҙa¶аУтИЪәПө°°Ч); payload(”yҺ§ДҝҳЛCARЮD»щТт, Изҝ№CD20 CAR)ЎЈЮDИҫ16РЎ•rәујУИлә¬15 U/mL DenaraseЈЁТ»·NәЛЛбГёЈ©өДРВхrЕарB»щЈ¬ҪөҪвҡҲБфDNA/RNAТФҪөөНХі¶ИІўМбёЯЭdуwјғ¶ИЎЈҙОИХКХ«@ЙПЗеЈ¬УГкҺлxЧУҪ»“QҢУОцЈЁMustang QЙ«ЧVЦщЈ©іхІҪјғ»ҜЈ¬НЁЯ^ЗРПтБчЯ^һVқвҝsІўЦГ“QҫҸӣ_ТәЈ¬НЁЯ^0.2 μm PESһVДӨіэҫъЯ^һVЈ¬Чоәу·ЦСbАдғцЧоҪKІЎ¶ҫЭdуw®aЖ·ЈЁVVPsЈ©ЎЈ
3.2 Particle bindingЈә
PBMCsЕcІЎ¶ҫоwБЈФЪКТңШПВ№ІЕарB1-4 hЈ¬ГҝәБЙэІЎ¶ҫҢҰ‘Ә2E7ӮҖјҡ°ыЈ¬ёРИҫҸН”өЈЁТІҫНКЗMOIЈ©һй2-10ЎЈК№УГanti-cocal antibodyНЁЯ^БчКҪФu№АІЎ¶ҫоwБЈ/Tјҡ°ыҪYәПЎЈЯ@АпөДcocalКЗұнЯ_ФЪVVPsІЎ¶ҫоwБЈұнГжөДМЗө°°ЧЈ¬ІЎ¶ҫоwБЈЕcјҡ°ы№І·хУэәу¶М•rйgғИЈЁ1-4РЎ•rЈ©ЯMРРөДҷzңyЎЈЯ@ӮҖ•rйgьcЦчТӘ°lЙъөДКЗІЎ¶ҫоwБЈЕcјҡ°ыұнГжөДМШ®җРФҪYәПЈЁНЁЯ^MDFө°°ЧЈ©Ј¬ЯҖҒнІ»ј°°lЙъёЯР§өДІЎ¶ҫЮDҢ§әНјҡ°ығИ»щТтұнЯ_ЎЈҙЛ•rҷzңyөҪөДcocalРЕМ–ҺЧәхНкИ«ҒнФҙУЪёҪЦшФЪTјҡ°ыұнГжөДНкХыІЎ¶ҫоwБЈЙПөДcocal°ьДӨМЗө°°ЧЈ¬Іў·ЗҒнФҙУЪTјҡ°ығИІҝұнЯ_»т®aЙъөДcocalө°°ЧЈЁTјҡ°ыұҫЙнІ»ұнЯ_cocalЈ©ЎЈ
3.3 In vitro PBMC transductionЈә
ІЎ¶ҫоwБЈЦұҪУјУИлөҪPBMCsЦРЈ¬°ҙХХГҝәБЙэҢҰ‘Ә2E6ӮҖPNMCsјҡ°ыЎЈ·Ц„eФЪөЪ3МмәНөЪ7МмУГБчКҪҷzңyјӨ»оәНЮDҢ§ЗйӣrЎЈ
3.4 In vitro PBMC transductionЈә
ИЎ5E4ӮҖCAR-Tјҡ°ыЈ¬К№УГCellTrace VioletҹЙ№вИҫБПЈЁЯ@·NИҫБПҝЙҙ©Нёјҡ°ыДӨЈ¬Еc°ығИө°°ЧЩ|№ІғrҪYәПЈ¬УГУЪЧ·Ыҷјҡ°ы·ЦБСЎЈ¶шЗТјҡ°ыГҝ·ЦБСТ»ҙОЈ¬ҹЙ№вҸҠ¶Иңp°лЈ©ЯMРРҳЛУӣЎЈҢўҳЛУӣәуөДCAR-Tјҡ°ыЕc5E4ӮҖNalm6Д[Бцјҡ°ы№ІЕарBЎЈЕарBуwПөК№УГX-VIVOҹoСӘЗеЕарB»щЈЁІ»ә¬IL-2,ТФЕЕіэНвФҙјҡ°ыТтЧУҢҰФцЦіөДёЙ”_Ј©ЎЈ5МмәуНЁЯ^БчКҪҷzңyҙж»оөДCAR+јҡ°ыЈЁНЁЯ^ЛА»оИҫБПЕЕіэЛАНцјҡ°ыЈ¬ФЩНЁЯ^CARМШ®җРФҝ№уwҙ_ХJЮDҢ§іЙ№ҰЈ©Ј¬ТФј°НЁЯ^CellTrace VioletҹЙ№вҸҠ¶ИөДПЎбҢіМ¶ИЈ¬ҹЙ№вҸҠ¶ИФҪөНЈ¬ХfГчјҡ°ы·ЦБСҙО”өФҪ¶аЎЈЯ@ҳУҪY№ыУГУЪБҝ»ҜCAR-Tјҡ°ыФЪД[Бцҝ№ФӯҙМјӨПВөДФцЦіДЬБҰЎЈ
3.5 PBMC-humanized mouse modelЈә
ҙЖРФNod.Cg-PrkdcscidIL2rgtm1Wjl/SzJРЎКуЈЁЯ@ӮҖРЎКуЦШ¶ИГвТЯИұПЭҢ§ЦВT/Bјҡ°ыИұК§Ј»IL2rg»щТтЗГіэИұ·ҰNKјҡ°ыЈ»MHC I/IIлpЗГіэЈ©ЎЈDay -4Ј¬НЁЯ^ ОІмoГ}ЧўЙд 2.5E5ӮҖ Nalm6јҡ°ыЎЈDay -1Ј¬УГ IVISңyБҝРЎКуД[БцШ“әЙЎЈёщ“юД[БцҙуРЎлSҷC·ЦҪMЈ¬ҙ_ұЈҪMйg»щҫҖТ»ЦВЎЈНЁЯ^ё№З»ЧўЙд2E7ӮҖPBMCsЎЈDay 0Ј¬ё№З»ЧўЙд”yҺ§CAR»щТтөД VivoVecІЎ¶ҫоwБЈЈЁ„©Бҝёщ“ю·ЦҪMХ{ХыЈ©Ј¬уw·e200 μLЎЈұOңyЦёҳЛД[БцШ“әЙәНСӘТәЦРөДCAR Tјҡ°ыЙъіЙј°ГвТЯјҡ°ыЧғ»ҜЎЈЯ@АпЧчХЯУГё№З»ЧўЙдPBMCөДФӯТтКЗКІГҙЈҝё№З»ғИә¬ҙуҫWДӨЎўДcПөДӨБЬ°НҪYөИҙОјүБЬ°НЖч№ЩЈ¬КЗГвТЯјҡ°ыҡwіІәНјӨ»оөДАнПлҲцЛщЎЈЧўИлё№З»өДPBMCsҝЙНЁЯ^ ё№ДӨБЬ°Н№ЬЯMИлСӯӯhПөҪyЈЁРи2-5МмЈ©ЎЈҢҚтһФOУӢЦРЈ¬Day -1ЧўЙдPBMCsәуЈ¬Day 0ҪoУиVivoVecЈ¬оAБфБЛTјҡ°ыЯwТЖЦББЬ°НҪMҝ—өД•rйgЎЈө«КЗИз№ымoГ}ЧўЙдЈ¬PBMCsҝЙДЬңюБф·ОІҝ»тЖўЕKЈ¬ё№З»ЧўЙд„tМṩёь·Җ¶ЁөДҫЦІҝОўӯhҫіЎЈ
3.6 NHP modelЈә
Ф“ҢҚтһҢўҫҺҙaҝ№CD20 CARөДVivoVecЭdуwоwБЈЈЁVVPsЈ©ЦұҪУЧўЙдөҪШiОІ«JәпөДБЬ°НҪYғИЈЁЧо¶а4ӮҖЈ¬ГҝМҺ≤1mLЈ©ЎЈГҝИХұOңyЈ¬Уӣдӣ„УОпөДуwңШЎў»о„У о‘BЎўКіУыЎўјSұгЗйӣrәНХыуwҪЎҝө оӣrЎЈҢҚтһҮАёсЧсКШ„УОпӮҗАнТҺ·¶Ј¬ГҝИХұOңy„УОпЙъАн о‘BЈ¬ІўФЪМШ¶Ё•rйgьcІЙСӘЯMРРБчКҪ·ЦОцЈЁҷzңyCAR Tјҡ°ыЙъіЙЎўГвТЯјҡ°ыұнРНөИЈ©ЎўDNAҷzңyЈЁУГУЪәуАmөДЙъОп·ЦІјҷzңyЈ¬ИзddPCRҷzңyЭdуw»щТтҪMЈ©ј°ЕRҙІЙъ»ҜҷzтһЈЁұOңyСӘіЈТҺЎўЙъ»ҜЦёҳЛөИЈ¬Фu№Ақ“ФЪ¶ҫРФ»тёұЧчУГЈ¬ИзCRSПакPЦёҳЛCRPЎўIL-6ЎўиFө°°ЧЎўёОГёALT/ASTөИЈ©Ј¬ТФФu№АуwғИCAR Tјҡ°ыөДЙъіЙЎў№ҰДЬЎўBјҡ°ыЗеіэР§№ыТФј°°ІИ«РФЎЈҢҰіц¬FCRS/ICANSЫEПуөД„УОпЈ¬°ҙ·Ҫ°ёК№УГБЛНРЦйҶОҝ№Ўў°ўДЗ°ЧңюЛШЎўөШОчгъЯMРРЦОҜҹЈ¬ЖдЦРТ»Ц»„УОпЯҖК№УГБЛөШИыГЧЛЙЎЈ
3.7 MDF sequenceЈә


- mIHCјјРgФЪҪвҙaД[БцОўӯhҫіј°ҳЛЦҫОпҷzңyЦРөД‘ӘУГј°ғһ„Э
- ҷzңyјӨЛШөДELISAФҮ„©әРоҗРНЎўМШьcј°ФӯАн
- ГАөВВ•іЈТҺәНИ«ГжПЎУРСӘРНФҮ„©ЯxРНЦёДП
- ДӨРФДIІЎАыНЧОфҶОҝ№ДНЛҺөДФӯТтј°ҢҰІЯ
- PRO-C6іЙһйоAңyCOVID-19ЦШ°Y»јХЯЛАНцпLлUөДкPжIСӘЗеҳЛЦҫОп
- З°СШЛЩЯf ЈәPRO-C6»тКЗCOL6-RDұнРН®җЩ|РФлyо}өДЖЖҫЦкPжI
- ҢҚтһРФЧФЙнГвТЯРФДXј№ЛиСЧ(EAE)ДЈРНҝ№ФӯЯx“сИ«№ҘВФ
- ГвТЯҪM»ҜФЪІЎАнФ\”аЦРөДЕRҙІ‘ӘУГғrЦөЕcТвБx
- СРЙъЙъОпНЖіцҙуКуPPAR-ҰГ ELISAФҮ„©әРДкҪKМШ»Э»о„У
- ГАөВВ•НЖіцРВЖ·°ІАКҢҷОпәНРуДБ„УОпФ\”аФҮ„©
- Э»ФҮШiХіө°°Ч/ХіТәЛШ5BГёВ“ГвТЯФҮ„©әРПЮ•r85ХЫғһ»Э
- AbsinДкҪKҝсҡgЩҸ¶аЦШғһ»Э,қMо~ЩӣiPhone,әДІДГвЩMЛН
- СРЙъҙЩдNЈәЪ…»Ҝө°°ЧELISAФҮ„©әРқM10әР85ХЫІўЛНМЧСb
- Э»ФҮЙъОпДкҪKғһ»ЭЈәјғҫGЗаГ№PCRЯMҝЪФҮ„©әРқM¶юЛНТ»
- ¶ЪELISAФҮ„©әРіГ¬FФЪ,җЫұШРЕИ«ҫҖ®aЖ·ЩIЩӣҒнТuҝЙҜBјУ
- СРЙъҙуКу»оРФСхҙШЈЁROSЈ©ELISAҷzңyФҮ„©әР8ХЫғһ»Э